Someone recently asked me “How do SPRI beads work” and I realised I was not completely sure so I went to find out.
My lab uses kits. Lots and lots of kits; kits for DNA extraction, kits for PCR, kits for NGS library prep and kits for sequencing. Kits rock! But understanding what is going on at each stage of the protocol provided with the kit really helps with troubleshooting and modification. How many people have added 24ml of 100% Ethanol to a bottle of Qiagen’s PE buffer without stopping to ask what is already in the bottle? The contents of the PE bottle are * answer at the bottom of this post.
I hope this post helps you understand SPRI a bit better and think of novel ways to use it. I’d recommend reading A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries in Genome Biology 2011 for the with-bead methods discussed at the bottom of this post. I am sure we’ll all be using their method a lot more in the future!
You are most likely to come across SPRI beads labeled as Ampure XP from Beckman.
First some handling tips:
- Vortex beads before use.
- Store beads at the correct temperature.
- Allow enough time for beads to come to room temperature.
- Pipetting is critical; use very careful procedures (SOPs) and well calibrated pipettes.
 How do SPRI beads work?
How do SPRI beads work?

Solid Phase Reversible Immobilisation beads were developed at the Whitehead Institute (DeAngelis et al 1995) for purification of PCR amplified colonies in the DNA sequencing group. SPRI beads are paramagnetic (magnetic only in a magnetic field) and this prevents them from clumping and falling out of solution. Each bead is made of polystyrene surrounded by a layer of magnetite, which is coated with carboxyl molecules. It is these that reversibly bind DNA in the presence of the “crowding agent” polyethylene glycol (PEG) and salt (20% PEG, 2.5M NaCl is the magic mix). PEG causes the negatively-charged DNA to bind with the carboxyl groups on the bead surface. As the immobilisation is dependent on the concentration of PEG and salt in the reaction, the volumetric ratio of beads to DNA is critical.
SPRI is great for low concentration DNA cleanup that is why it is used in so many kits. The reagents are easy to handle and a user can process 96 samples very easily in a standard plate. Alternatively the protocol can be easily automated and tens or hundreds of plates can be run on a robot in a working day. The binding capacity of SPRI beads is huge. 1ul of AmpureXP will bind over 3ug DNA.
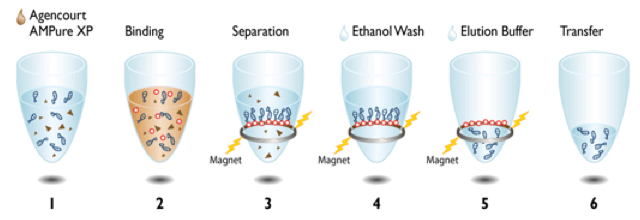 |
| This is the typical SPRI protocol from Beckmans website. |
Size-selection with SPRI:
Again the concentration of PEG in the solution is critical in size-selection protocols so it can help to increase the volume of DNA you are working with by adding 10mM Tris-HCL pH 8 buffer or H2O to make the pipetting easier. The size of fragments eluted from the beads (or that bind in the first place) is determined by the concentration of PEG, and this in turn is determined by the mix of DNA and beads. A 50ul DNA sample plus 50ul of beads will give a SPRI:DNA ratio of 1, as will 5ul pipetting (but much harder to get right). As this ratio is changed the length of fragments binding and/or left in solution also changes, the lower the ratio of SPRI:DNA the higher larger the final fragments will be at elution. Smaller fragments are retained in the buffer usually discarded and so you can get different size ranges from a single sample with multiple purifications. Part of the reason for this effect is that DNA fragment size affects the total charge per molecule with larger DNAs having larger charges; this promotes their electrostatic interaction with the beads and displaces smaller DNA fragments
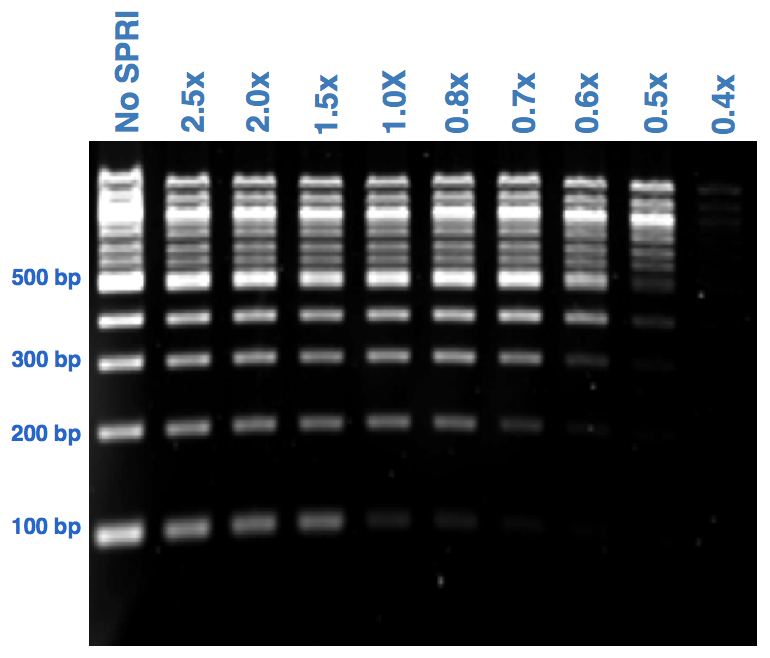 |
| SPRI size selection from Broad “boot camp” |
 |
| SPRI size selection from Broad “boot camp” |
Broad Boot Camp http://www.broadinstitute.org/scientific-community/science/platforms/genome-sequencing/broadillumina-genome-analyzer-boot-camp
 With-bead SPRI cleanup:
With-bead SPRI cleanup:
An incredibly neat modification of SPRI clean-up is the “with-bead†method developed by Fisher et al in the 2011 Genome Biology paper. Rather than using SPRI to clean-up discrete steps in a protocol these are integrated into a single reaction tube method, thus reducing the number of liquid transfer steps. After each step DNA is bound to beads by addition of the 20% PEG, 2.5M NaCl buffer, washes are performed as normal with 70% ethanol but the DNA is not eluted and transferred. Rather the master-mix for the next step in the protocol is added directly to the tube. The final DNA product is eluted from beads for further processing e.g. Illumina sequencing. This modification increases DNA yields in Illumina library prep as multiple transfer steps are removed, reducing the amount of DNA lost at each transfer.
How will you use SPRI in your lab?
PS: the Qiagen PE bottle contains 6ml of water.
PPS: If DNA and Carboxyl are both negatively charged what is happening on a SPRI bead? There appears to be a bit of a black hole in the literature I read through about how the SPRI beads actually work at the molecular level. Digging around some more it appears that the PEG can induce a coil-to-globule state change in the DNA with the NaCl helping to reduce the dielectric constant of the solvent, PEG also acts as a charge-shield. These mechanisms may be behind the “crowding” talked about on other sites. Perhaps someone out there can enlighten me?
Refs:
Lis JT. Size fractionation of double-stranded DNA by precipitation with polyethylene glycol Methds Ezymol (1975)
Lis J. Fractionation of DNA fragments by polyethylene glycol induced precipitation. Methds Ezymol (1980)
DeAngelis et al. Solid-phase reversible immobilization for the isolation of PCR products. NAR (1995)
Hawkins et al. DNA purification and isolation using a solid-phase. NAR (1994)






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Thanks for the insight. I have a question about the mechanism of DNA binding by the beads. You mentioned carboxyl molecules. Carboxyl group is negative once dissociated from the proton, so how does it bind DNA, which is also negative?
It turns out that by answering one person's question I have inevitably generated other questions along the way. I have updated the end of the blog to try and start answering the "negative charge" questions. However I don't understand the chemistry at the molecular level and am hoping someone more knowledgeable can help.
nice post. informative.
The carboxyl group is "activated" in the presence of acid solution—a hydrogen on the carboxyl group becomes positively charged thus attracting the negatively-charged DNA.
To be longer-winded, I think Anonymous is suggesting that at a low enough pH the carboxyl group is protonated, not ionised, and that with the electronegativity of two adjacent oxygen atoms it is showing an induced positive charge (not a full positive charge like a proton or sodium ion would carry, but partial positive charge due to the electrons from the O-H sigma bond hanging out around the oxygen nuclei more of the time). Seems reasonable, depending on the pH, and assuming that the phophoryl groups of the DNA backbone remain deprotonated whilst the carboxyl groups of the beads are (mostly) protonated. Anyone know the pKa values??
James, I don't suppose you could tell us what (if any!) the difference in buffer is for Sanger sequencing reaction cleanup (ie: ssDNA of 25-1000 bases) ?
The pKA of a carbonyl group RCOOH is ~15ish. Could this work by a chaotropic effect?
Negative carboxyl-positive salt-negative DNA
Does anyone know what the difference is between RNA and DNA selective SPRI beads?
RNA are quaranteed RNAse free
To my mind that's exactly what I refer to a lovely blog article! Do you utilize this site for your private joy solely or you still exploit it profit wise?
Excellent blog you have here. I’m a long time reader, but haven’t commented before; I just had to say how much I enjoy the site. Where did you get this Wardress theme? Is it custom? I really like it next-generation sequencing and microarray technologies.
The addition of PEG leads to "depletion" effect, i.e., the PEG molecules push DNA aggregating. The high NaCl concentration helps this effect ("salting-out"). This salting-out and depletion effects promote the binding of the carboxylic groups on beads surface with DNA via hydrogen bonding.
Hi James@cancer,
I loved reading this piece! Well written!
Merlen Hogg
Tormod Helleland
You wouldn't happen to know what molecular weight the PEG is would you? Or does that not matter?
My lab uses kits. Lots and lots of kits
And it shows.
PS: the Qiagen PE bottle contains 6ml of water
False! As as scientist, you should attempt to do at least minimal experimentation before making any claims.
The Qiagen PE bottle contains 50 mM Tris pHed to 7.6 with HCl. Go ahead and test PE for pH, Cl- and secondary amines.
True, substituting Tris for water does not make that much of a difference but the residual protein contamination is definitely higher when unbuffered 80% ethanol is used.
Also true that it is completely retarded to buffer at 7.6 with Tris but with the Tris being molecular biologists' favorite reagent, one can hardl;y expect anything else…
Is there a method to recycling the beads?
Informative discussion!!
Can anyone please help tell me the reason of DNA loss while undergoing SPRI purification of the sheered DNA?
I use PEG 8000. I ligate adapters on the beads, and reuse the beads for following step with 20 % PEG (8.000), 2.5 M NaCl.
To my experience the binding capacity of the beads is as low as 30ng DNA/ul beads. You should perform some tests to figure out the capacity of the beads under your conditions.
The comments above assume that the DNA binds "magically" to the beads due to carboxylated coatings. This same myth runs with the use of silica as well. Bottom line is the carboxyls are on the surface to lightly repel the DNA off the surface once the DNA-bead aggregate is place in an aqueous solution that allows the DNA to fully hydrate back into solution phase.
The other reason for carboxyl groups is shield the DNA from coming in contact with sticky plastic surface (or silica)- i.e. use of nylon, for Southern blots or polystyrene for ELISA.
The process of DNA capture on a bead surface is the same as process of precipitating DNA for centrifugal collection. The long complex polymer of DNA remains in solution phase due to the structured water layer or cloud that forms around the DNA polymer primarily due to hydrophilic/ionic interaction of (deoxy)ribosugar-phosphate backbone of the nucleic acid. When "water hungry" alcohols or polyalcohols (PEG) are added to very high concentrations, the water cloud is disrupted, becomes significantly thinner, which results in the DNA transitioning from solution phase to a flocculated or precipitated phase. DNA transitions from a long polymer water cloud, coated polymer to water-stripped condensed "sticky ball". These DNA "balls" will stick to or aggregate with each other or to any surface that the ball collides into, surface such as that of a bead, fiber, or membrane. This flocculation process can be induced with adding salts to high concentrations, i.e. sodium perchlorate, ammonium acetate, lithium chloride, and with proteins the use of alcohols or high concentration of sulfate salts.
In reference to carboxyl and bead surface; if the surface was coated with amines, the DNA (via phosphate backbone) would adsorb to the surface via ionic bonds and not elute from bead surface. This ionic adsorption would occur with metal oxide surfaces including semi metal oxides such as silica if Lewis acid sites are exposed – which is common with most materials.
Nucleic acids have been isolated using plastic, silane, silica, ferrite, or even clay beads/particles. For all cases if the surface is pretreated with a starch, proteins or some other slightly negative charged coating, which shields the surface from DNA, the DNA easily elutes from the surface. Thus by coating the silica or plastic surface with carboxyl groups, the DNA when it transitions to fully hydrated form during elution phase will be ionically repelled by carboxyl coated surface.
Side note, smaller nucleic acid polymers are more soluble in these alcohol solutions due to their smaller size – form smaller condensed balls. Cold or time will allow the smaller balls to aggregate form larger balls and increase surface area of the ball to stick to a solid support or be spun out by centrifugation.
So I hope this helps in dismissing the myth that DNA has special ionic attraction to carboxyl or silicon oxide surface and that the process is due to physical chemistry of flocculation / precipitation process.
Additional comments to above tirade by the same author; Guanidinium salts (i.e. chloride or isothiocyanate salts) like alcohols, PEG, and acetate or sulfate salts, at high concentrations, with or without alcohol, cause DNA to precipitate / flocculate out of solution phase as well.
Look up the "Rene Boom" papers using this salt with silica based solid phase to isolate DNA.
Below is most cited with abstract- though look up the 1999 paper as well.
_____
Rapid and simple method for purification of nucleic acids.
Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J.
Source: J. Clinical Microbiology (1990) 28 (3) 495 – 503.
Department of Virology, Academic Medical Center, Amsterdam, The Netherlands.
Abstract
We have developed a simple, rapid, and reliable protocol for the small-scale purification of DNA and RNA from, e.g., human serum and urine. The method is based on the lysing and nuclease-inactivating properties of the chaotropic agent guanidinium thiocyanate together with the nucleic acid-binding properties of silica particles or diatoms in the presence of this agent. By using size-fractionated silica particles, nucleic acids (covalently closed circular, relaxed circular, and linear double-stranded DNA; single-stranded DNA; and rRNA) could be purified from 12 different specimens in less than 1 h and were recovered in the initial reaction vessel. Purified DNA (although significantly sheared) was a good substrate for restriction endonucleases and DNA ligase and was recovered with high yields (usually over 50%) from the picogram to the microgram level. Copurified rRNA was recovered almost undegraded. Substituting size-fractionated silica particles for diatoms (the fossilized cell walls of unicellular algae) allowed for the purification of microgram amounts of genomic DNA, plasmid DNA, and rRNA from cell-rich sources, as exemplified for pathogenic gram-negative bacteria. In this paper, we show representative experiments illustrating some characteristics of the procedure which may have wide application in clinical microbiology.
It is a big attractive blog.
ADP Assay Kit
Hi, thank you for the very informative blog!
I was wondering whether the initial buffer composition would affect binding of the DNA to the beads? I am planning to purify ChIP DNA material with SPRI beads but my elution buffer contains 1% SDS and 0.1M NaHCO3. Not sure if these conditions wouldn't be too harsh…
Thanks!
Please bear with me as this is entirely new to me. If I purchase the Agencourt AMPure XP kit, and I need to retain DNA >600 but definitely eliminate DNA <200 then I can simply alter the prescribed 1.8 X Reaction Volume so that it is, according to the gel image above, 0.5X ?? Can I then reuse the beads for additional reactions? I see PEG mentioned above, is that contained in the AMPure mix? If I can reuse the beads, what do I resuspend them in to recreate the original bead mixture?
Thanks again for your patience. By the way, this post has helped me come close to understanding what is going on. Thanks.
Hi! I use AMpure beads to clean-up my ChIPseq libraries and use the Resuspension Buffer from BIOO Scientific's Nextflex ChIPseq-kit. Anyone who has an idea of what that contains or what I could use instead?
Thanks!
Hi, I was planning to try the same. Did you already try to purify your ChIP samples with the beads. Did it work?
you describe about this topic really well thankssigma antibody
Great post, thank you for all the info! I was wondering if you had a general idea of the ratio of 20% PEG solution to beads in a with-bead library prep? We are wanting to try this with Agilent SureSelect library prep, which uses a post-shear bead volume of 180ul. Thank you for any insights!
Nice start guys…I went through the website and I found that you made a decent point here. Keep up the topic that everyone can choose one of the best. Thanks.Sigma antibody
The pKa of tris is ~8. I was taught that a buffering reagent is generally effective at ±1 the pKa. Ie tris is suitable for 7-9.
why is it "retarded" to buffer at 7.6 with tris?
What makes the aggregated DNA prefer the beads versus the surface of the container the solution is in? Or is there no preference and we merely exploit that the beads have a much larger surface area than the container?
I found your blog when I was looking for a different sort of information but I was very happy and glad to read through your blog. The information available here is great. . I know something information, to know you can click here
water softener
water filter
has any tested this for genomic DNA purification from tissue? any recommendations?
The working solution is pH 8, so the carboxylate will have a negative charge.
If SPRI beads are charged is there the possibility of any problems with pipetting with conductive tips for example in automated workstations?
What would you suggest when you want to focus on smaller fragments? Since the beads are usually washed with 70-80% alcohol it seems to be more likely to lose the small fragments if I understood your side not correctly. When you say "cold or time" would help, were you referring to the DNA binding condition? How cold is "cold"? And in what way can I get rid of the large fragments but keep (only) the small ones?
Thanks
I had been trying to remove the Illumina adapters ~120bp from my PCR amplified product. At 0.7:1 Ampure beads ratio I am still unable to remove the 120bp fragments. Is it necessary to add additional PEG solution and if so what is the % of PEG to add?
Great piece of writing, thank you for taking the time to be so thorough.
Had no idea what a SPRI bead was before reading this. Now I know. Thanks!
-Jackie @ construction site cleanup
I read that Post and got it fine and informative. Please share more like that…
office cleaning richmond
Thank you for your post. This is excellent information. It is amazing and wonderful to visit your site.
metabolic dna testing
You get better adapter-dimer removal by doing two rounds of clean-up than reducing the bead concentration.
You can also try titrating the adapters down. That way, there's less chance of creating dimers in the first place.
Very good post. I enjoy to read this post. Office Cleaning Services
Anonymous23 May 2014 at 08:13
What would you suggest when you want to focus on smaller fragments? Since the beads are usually washed with 70-80% alcohol it seems to be more likely to lose the small fragments if I understood your side not correctly. When you say "cold or time" would help, were you referring to the DNA binding condition? How cold is "cold"? And in what way can I get rid of the large fragments but keep (only) the small ones?
Thanks
I found your blog when I was looking for a different sort of information but I was very happy and glad to read through your blog. The information available here is great. . I know something information, to know you can click hereRetial cleaning services melbourne
I found your blog when I was looking for a different sort of information but I was very happy and glad to read through your blog. The information available here is great. . I know something information, to know you can click hereRetial cleaning services melbourne
thanks, great post! love getting into the basics
Hi Laura, sometimes we forget that we ever learned them at all!
Thanks for sharing
Does anyone know if using 15,000-20,000 PEG will work the same way as using 8000 PEG? I notice on the 15k-20k PEG it says that it's made using M.W. 7000-9000 PEG that is internally joined by an aromatic ring, so I wonder if the two arms of 7k-9k avg polymer will work the same…
Hello!
You have very interesting blog with winderful posts, thank you for creating this all and sharing with us 🙂
I learned alot on the topic of how do SPRI beads work, thank you! Best wishes
[…] DNA is then purified with SPRI Select (read my 2012 “How SPRI works” post – over 150,000 other people […]
[…] Be meticulous about your bead cleanups (read about how SPRI-beads work here) […]
[…] paper was published in Nature in 2012 and has 2076 citations. The piece I wrote the same year, “How do SPRI beads work?”, has had over 150,000 page views! Last year I moved across to this […]
Almost in all the homemade recipe of SPRI beads, there mentioned the adding 0.05% Tween 20. However, as soon as you adding the Tween 20 (or other detergents like Triton X-100, NP-40, SDS, etc.), the whole reagents get viscous. And when you try to transfer the reagents with micropipettor, it is easy to stick to the tips, while AMPure XP is not like that.
Has that ever happed to you?
Nice article.
Do you have experience of other beads such as Celemics? (www.celemics.com)
due to the instruction of the SPRI beads, the concentration of NaCl cause no influence in the selection of DNA bp region. I guess the concentration of NaCl might not be the key reason of the selection.
Found this useful to explain how PEG reversibly bind to DNA
https://blog.genohub.com/2014/05/07/peg-size-selection-and-precipitation-of-dna-libraries-how-ampure-or-spriselect-works/
Thanks for the tip.
Hi, does anyone can help us ?? We have a protocol for Clean-up with Bead Size Selection:, and it says we need 100ul to start the reaction. Do you know what is a good concentration of DNA to start de BED SIZE SELECTION?? ( we want to keep fragments for 200-500)?? Thank you all in advance
Very informative article.
All the beads in the market are bad at small DNA/RNA under 100 bases. We have developed a beads for DNA/RNA as short as 20 bases:
https://biodynami.com/product/spri-beads-microrna-oligo-purification/
It works, but the mechanism is not clear.
[…] który wzmacnia i stabilizuje wiązanie DNA /RNA do złoża jest roztwór 20% PEGu w 2,5M NaCL. To od stężenia tego sekretnego medium zależy powinowactwo kulek do określonej długości fragmen…. To właśnie wolumetryczna manipulacja końcowym stężeniem PEGu decyduje o tym co i jak zwiąże […]