About two years ago when we started using Fluidigm Access Array sequencing we noticed something in the reads that was a bit of a surprise, although not totally unexpected once we realised what was going on. We were amplifying all the exons in seven cancer genes across 48 cancer cell lines, sequencing them in a single experiment and finding known SNPs at the expected allelic ratios. However we also found quite a large number of what seemed to be random deletions and truncations in the targeted regions, and these all occurred towards the beginning of the reads.
One of the “problems†with many amplicon sequencing methods is that they often tail locus specific primers with NGS adapters and this means you have to read through the PCR primer before you get to interesting results in your samples. In our case the first 25bp or so of each read was from the primer.

It appeared that we were seeing the incorrect by-products of the oligo production process. Oligo manufacturers use varying methods of clean-up and QC but none are perfect and it looks to me like NGS might be the ultimate, if slightly expensive, oligo QC tool.
One way to test this would be to get the same pair of oligos made by multiple companies, PCR amplify a control DNA template with all of them and then sequence the primer sequence only in a pooled sequencing run. Anyone fancy collaborating?
An oligo “primerâ€: I thought I would follow up on this with an overview of oligo manufacture and some tips for PCR primer design.
You can buy oligos from many places and a very few labs still make their own. It is possible to get oligos of up to 400bp in length, chimeric oligos (DNA:RNA) and with all sorts of modifications: Fluroescent dyes, Amino-modified, Biotinylated, Phosphorylated, 2′-Deoxyinosine (INO) 5-Methyl-dC, Phosphorothioate, dI, dU, 2′-Deoxyuridine (URI), Amino C6-dT, Spacers (Deoxyabasic or C3), Thiols, etc, etc, etc. Most people are simply out to buy a primer for PCR or Sanger sequencing but there are also RNA, siRNAs, PNA, LNA and many other types available for many applications.
Choosing a provider most often comes down to cost and the price per base is very low for a standard desalted PCR primer. However the options offered by oligo manufacturers are numerous and some might be a prerequisite for your experiment.
Most standard oligos are supplied already quantified and even at your pre-determined concentration. The amount of oligo you actually get is dependent on the synthesis scale (how much is made), the efficiency of each coupling and the purification used. The lowest synthesis scale is generally fine for PCR applications, however if you want specific cleanup of your oligo you may have to get a larger scale synthesised. Most providers suggest that you resuspend oligos in TE rather than water, which can be slightly acidic. I have always used a buffer with a lower amount of EDTA (10 mM Tris, pH 8.0, 0.1 mM EDTA) as this can inhibit some molecular biology applications at higher concentrations.
Oligo QC: The current standard for QC is mass spectrometry (MALDI-TOF or ESI-MS), or gel electrophoresis (CE or PAGE). MALDI-TOF is used for most applications because of its high throughput, but ESI is better for oligos >50bp.
Oligo Yields: Most companies will specify yields that seem incredibly low compared to the starting nucleotide concentrations. The oligo synthesis is performed sequentially with a single nucleotide being coupled to the growing oligo in a 3’ to 5’ direction. This coupling is often less than 99% efficient so some of the oligos are not extended. This means the final product is a mix of full-length product (n) and truncated sequences (n-1, etc).
Oligo purification can be performed in many ways, cartridge, HPLC, PAGE. On the Sigma website there is a very handy table showing which clean-up you should choose for different applications. 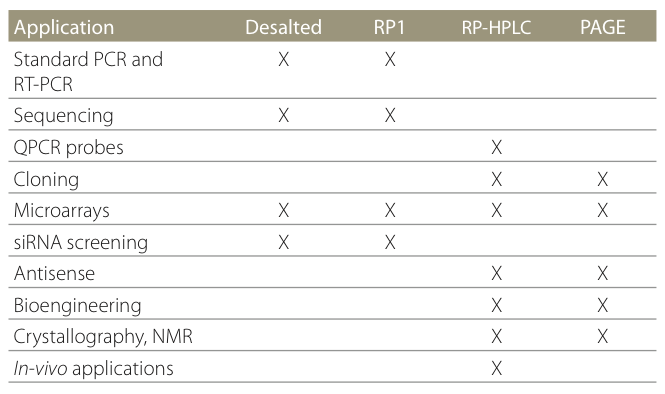
Sigma’s clean-up guide
Desalting: This is the most common and cheapest clean-up method and is perfectly fine for standard PCR based applications. Desalting removes by-products from the oligo manufacturing process. If your oligos are >35bp then desalting will not remove the relatively high number of n-1 and other truncated oligos.
Cartridge: A reverse-phase cartridge purification by Sigma will give about 80-90% yield. Full length oligos contain a 5′-DMT and are separated from truncated sequences by making use of their higher hydrophobicitiy. However this difference is reduced as oligos increase in length and should not be used for anything >50bp.
HPLC: A reverse-phase HPLC purification allows higher resolution and gives higher yields of >90%. HPLC also allows purification of larger amounts of oligo (>1 umol). Again this method is not ideal if oligos are more than 50bp.
PAGE: A poly-acrylamide gel electrophoresis can achieve single-base resolution and very high-quality purification and is highly recommended for longer oligos, >50bp. Unfortunately the yield after gel extraction can be quite low.
Primer design tips: Oilgos for PCR, qPCR and sequencing are pretty easy to design if you follow a few simple rules. Once you have primers designed it pays to use the Blat and in silico PCR tools on the UCSC genome browser. Order the lowest synthesis scale for basic PCR applications and resuspend oligos in low EDTA TE (10 mM Tris, pH 8.0, 0.1 mM EDTA). If you are in any doubt about contamination, throw oligos away and buy new ones!
- Use Primer 3: Primer 3 is the tool many others are based on, forget all the other rules just use it! But in the spirit of educating readers here are the other rules…
- Amplicon length: Standard PCR is fine up to 1-3kb, over this then the primer design may not need to change but the reaction conditions almost certainly will. For qPCR keep amplicons around 100-200bp.
- Oligo length: 18-22bp is optimal and should allow high specificity and good annealing characteristics.Longer tehn 3-0bp and you can affect teh annealing time reducing reaction efficiency.
- Melting Temperature: A Tm of 52-58C works best in most applications, make sure primer pairs are as similar to each other as possible and test using a gradient cycler run.
- GC Content: The GC content (the number of G’s and C’s in the primer as a percentage of the total bases) of primer should be 40-60%.
- Avoid secondary structure: If your primer develops secondary structures you will get low or even no yield of the final product. 2’ structure affects primer template annealing and thus the amplification. Internal hairpins and primer dimers are common problems.
- Avoid repeats and runs: Repeats (e.g. di-nucleotides) and mono-nucleotide runs can easily misprime (and promote 2’ structure), try to avoid them or keep to fewer than 4-6bp.
- Don’t design to homologous regions: Sounds obvious but if your primer can anneal to multiple regions in the genome then you are likely to get multiple products in your reaction. Blast or Blat your final sequences before ordering.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
So what did you end up ordering for NGS amplicon sequencing? PAGE?
As long as the primers are amplifying the targeted region, everything is fine. If errors lead to unspecific amplification your read yield decreases. But if this happens at below 1 to 5 % it would probably not hurt too much. Do you have numbers on that?
We ordered very standard HPLC purified oligos. The errors are few enough not to warrant the expense and time in getting PAGE purified. Some of the reads do start one or two bases off predicted and the oligos do contain errors.
specialize in amino acid analysis and oligo synthesis
Thank you so much for provide the relevant information about Polymerase Chain Reaction and primer.Its awesome information !