A recent BioRxiv report from the Gerlinger group at ICR describes a targeted ctDNA sequencing method that uses error correcting UMIs to achieve 100% sensitivity for mutant allele frequencies of >0.15%, and 87% at >0.075%, and reduce false-positive mutation calls by 98.6%, without adversely affecting the false-negative rate.

Circulating tumour DNA (ctDNA) sequencing is revolutionising research and treatment of cancer patients, albeit a little more slowly than the impact foetal cfDNA analysis is having on NIPT. Both methods rely on analysis of the cell free portion of DNA in blood plasma – if carefully handled and extracted cell-free DNA can be allow minimally invasive, “liquid biopsy” testing of cancer patients, and detects low-frequency variants allowing earlier detection of response to or failure of treatment, emergence of resistance, and tumour evolution analysis. A major problem facing users of ctDNA for low-frequency mutation detection is the need for high specificity and sensitivity, and as detection limits are pushed well below 1% mutant allele frequency (MAF) a common problem is noise in the library prep and seqeuncing methods.
The report: “Combined Mutation Detection and Copy Number Profiling by Error-Corrected ctDNA Sequencing” by uses Agilent’s latest exome library prep chemistry “XT HS” which incorporates unique molecular indexes (UMIs), or molecular barcodes (MBCs) as Agilent refers to them, which I am sure are going to be critical for pushing the boundaries of low frequency allele detection. 4ml of blood plasma was processed using Qiagen QIAmp circulating nucleic acid kits to yield around 25ng of ctDNA for analysis.
The group state how their use of of error correcting UMIs significantly improves low-frequency mutant allele detection sensitivity, whilst also dramatically reducing false-positive rates. They describe copy number analysis using the off-target reads to detect chromosome arm and/or major gene amplifications. And they discuss the impact of “clonal heamatopoiesis” on TP53 mutation calling point to refs 22&23 making a clear recommendation that blood cells are sequenced in cancer patients to show that TP53 variants are coming from the tumour.
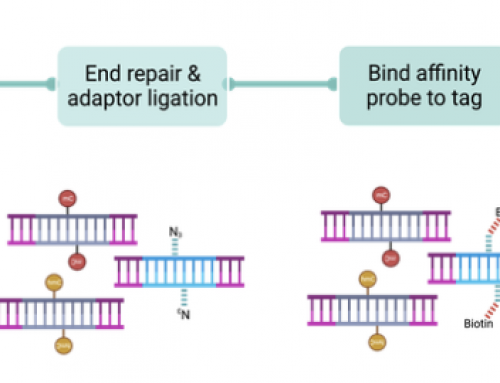
How do UMIs work to reduce error rates
The use of UMIs is described in a recent review from Nitzan Rosenfeld’s group here at CRUK’s Cambridge Institute: Wan et al 2017 Nature Reviews Cancer. Importantly Jonathan and colleagues point to the use of replicate library prep and sequencing as being important in reducing error rates, I expect a lot of future work will combine all the error correction possible to get to ultra-low VAFs.
 UMI methods add random stings of bases to DNA molecules such that the random string can be used to remove hybridisation capture and PCR biases, as well as allowing removal of random sequencing errors. Mutations are generally called at a specific threshold of UMIs – variants with only a single, or very few, UMIs are either removed from analysis, or flagged as potential false positives in downstream analysis. Generally UMIs are added as part of the adapter sequence, and are read as part of the sample barcoding read.
UMI methods add random stings of bases to DNA molecules such that the random string can be used to remove hybridisation capture and PCR biases, as well as allowing removal of random sequencing errors. Mutations are generally called at a specific threshold of UMIs – variants with only a single, or very few, UMIs are either removed from analysis, or flagged as potential false positives in downstream analysis. Generally UMIs are added as part of the adapter sequence, and are read as part of the sample barcoding read.
The Gerlinger group sequenced their 28 samples to a median of 1782x coverage, they required three independent (different UMIs) reads for mutation calling making their limit of detection 0.17%. Mixing experiments were performed to test this using two samples with 16 homozygous SNPs in their small panel, sequenced to 21,000x coverage. Analysis was performed by calling variants based on UMIs or on genomics coordinates (another way to determine if a molecule is unique). After de-duplication the UMI data was 2400x whilst the co-ordinate data was only 1500x – this was primarily because UMIs tag molecules irrespective of whether they have the same co-ordinates by chance (something that will happen with high frequency in ctDNA panel sequencing), and also tag both strands with different UMIs. The 16 homozygous SNPS were recovered with 100% sensitivity at 0.15% AF, 87.5% at 0.075% AF and 68.75% at 0.0375% AF.
CNV calling in off target reads of a 136kb panel
The group used CNVkit to call genome-wide copy number form the off-target reads in their data. Only 71% of patient samples were amenable to this analysis. They detected chromosome arm loss (e.g. 17P in 19/20 patients) and gene amplification (e.g. ERBB2 in a single patient). I was a little disappointed on reading the description of CNV calling in the report. Whilst the results are good, they are limited to chromosome arm and/or major gene amplification detection. For many cancers we need better CNV analysis than this and I suspect the reason the results were so limited was the size of the panel. You simply don’t get enough of-target across the genome to do a robust genome-wide CNV analysis. I’d suggest pooling the pre-capture libraries for shallow whole genome sequencing.
Is affordable early detection in lung cancer possible?
Given that the Gerlinger report used 21,000x coverage in their limits of detection analysis and were “only” able to achieve 68.75% sensitivity at 0.0375% allele frequency it looks like the use of ctDNA sequencing as a screening assay is going to require further improvements. Ultimately the limit is the number of mutant molecules in the sample so assay improvements that convert closer to 100% of DNA molecules into sequencing reads will help push detection sensitivity limits down by at least an order of magnitude.
The detection sensitivity required for a screening assay is likely to be very disease specific. In diseases like lung cancer where we can easily identify which people to test might we already be at a level that has an impact on patient outcome? If the ctDNA assay can be run at low-cost then an annual screen for the common lung cancer mutations might detect a larger number of patients early enough for surgery? Perhaps this, could be the first screening application? Given the number of smokers, and the tax already added to cigarettes a test that costs £500 may be affordable for the government to commission – here’s my back of the envelope calculations:
- In the UK 17% of adults still smoke, and go through about half a pack per day.
- At £10 per pack that equates to around £2000 per year per smoker
- I’ll assume a test costs £500 (can we do a lung cancer assay at 21,000x at this price?)
- If we screen 10% of smokers then we’d need to add around 25% to the cost of a pack of cigarettes to cover the test costs.
- Detecting stage 1 lung cancer can mean >50% 5 year survival rates!
Summary
Agilent have arrange a Genomewebinar by Marco Gerlinger on December 5th 2017 at 6:00 pm: Marco will discuss the work presented in the paper (also in an Agilent whitepaper) and I am sure go into a bit more detail about the molecular barcode-based error correction methods.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
What about Qiaseq from Qiagen? Also, UMI enabled but significantly cheaper. However, its custom panel is only up to around 300 genes.
Sorry but I’ve not really looked deeply enough into Qiagen’s sequencer. The underlying chemistry as far as I am aware means it should not be affected in the same way as Illumina’s ExAmp clustering, however any pooled sequencing should be designed in a way that mixups like this can be identified and removed, or at least accounted for as a source of noise/error.