Last week I followed up on the index-swapping issue after Illumina released their white paper and also covered what Ethan Linck at The Molecular Ecologist had posted about the Sinha et al BioRxiv paper. In that post I said I’d write a follow-up post about index design over the weekend – here it is!
Single index sequencing is a really bad idea
Unique Dual Indexes
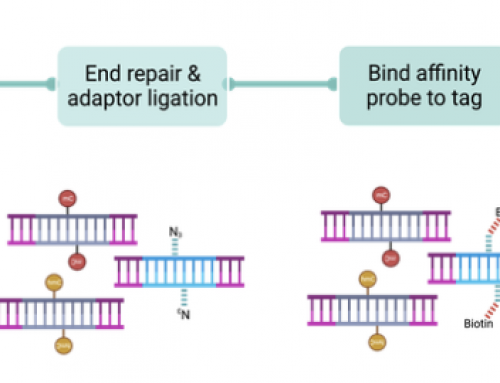
In their white paper Illumina used Unique Dual Index (UDI) combinations to detect and quantify index-swapping. A UDI is exactly what it says on the tin – both ends of the index combination are unique in the library pool. So for a 12×8 Nextera prep you could create 12 8-sample UDI pools by pooling across diagonal rows, but not nice and easily by pooling down or across rows (see figure below).

Illumina showed data from a 4 UDI pool where ~24% of the reads came from each of the UDI combinations, and 4% of reads were lost to the 12 possible incorrect pools due to index-swapping. Illumina calculated that this index-swapping led to about a 1:600 error rate in index assignment.
To make life easy for anyone running larger pooled experiments we’ll need Illumina and other companies to make these indexes for us. You can make UDI pools if you are using the standard Illumina indexing plates (or knock-offs from other manufacturers)…but you’ll need to carefully pool across diagonal rows. Or re-array your index combinations before use such that they can be pooled nice and easily in columns…but be careful not to contaminate them while pipetting! Use a robot if you can.
Redesigning NGS indexes
Assuming that Illumina are going to release a set of UDIs for TruSeq applications (don’t forget Nextera) then this represents an opportunity to consider other issues with index designs and come up with something far better than what we’ve got now. Simply fixing the index-swapping issue without considering some of the issues listed below would be a wasted opportunity.
Indexes need to be unique at both ends: goes without saying (if you were listening) we need to use paired-end indexes (Illumina still use single-indexes for the TruSeq smallRNA kits).
Indexes need to be unique across kits: some 6bp indexes can be found in some 8bp indexes, which can be found in some 10bp indexes (how did we end up in this mess). Index combination should not be reused.
Indexes need to be longer than 8bp: longer indexes are less prone to errors from sequencing, misincorporation during PCR (or even during oligo-synthesis).
Indexes could be MUCH longer than 8bp: Illumina’s sequencing quality score estimation makes use of the chastity filter over the first 25bp to remove low quality reads. A 25bp index read would mean q-scores on indexes should be more reliable. A 300 cycle kit has 25bp extra for indexing so the additional cost of a paired-25bp index might be as little as $100 on a $1500 lane. Seems a small price to pay to know your genomes/exomes/RNA-Seq samples were demultiplexed properly on NovaSeq S4.
Do know someone who can make oligos particularly well?







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Just an FYI — the index reads on instruments released after the NextSeq 500 only support index reads <= 20 cycles. Unclear whether that will ever change.
Another way to address this issue is to go back to inline indexes. That way people have more control over index compositions and read lengths.
Hi BTCL, I missed that change was it in the software for the machines or in BCLtoFASTQ? In-line is certainly a way to go. I know a lot of people are looking into this now it’s become a more openly discussed problem. Its a shame this discussion was not happening a year ago! Or that Illumina did not mention it when launching HiSeq 4000.