Multiplexing is the default option for most of the work being carried out in my lab, and it is one of the reasons Illumina has been so successful. Rather than the one-sample-per-lane we used to run when a GA1 generated only a few million reads per lane, we can now run a 24 sample RNA-seq experiment in one HiSeq 4000 lane and expect to get back 10-20M reads per sample. For almost anything other than genomes multiplexed sequencing is the norm.
But index sequencing can go wrong, and this can and does happen even before anything gets on the sequencer. We noticed that PhiX has been turning up in demultiplexed sample Fastq. PhiX does not carry a sample index index so something is going wrong! What’s happening? Is this a problem for indexing and multiplexing in general on NGS platforms? These were the questions I have recently been digging into after our move from HiSeq 2500 to HiSeq 4000. In this post I’ll describe what we’ve seen with mis-assignment of sample indexes to PhiX. And I’ll review some of the literature that clearly pointed out the issue – in particular I’ll refer to Jeff Hussmann’s PhD thesis from 2015.
The problem of index mis-assignment to PhiX can be safely ignored, or easily fixed (so you could stop reading now). But understanding it has made me realise that index mis-assignment between samples is an issue we don not know enough about – and that the tools we’re using may not be quote up to the job (but I’ll not cover this in depth in this post).

Issues with index mis-assignment and quality were initially noticed when we detected Illumina’s PhiX control in demultiplexed Fastq data. PhiX is supplied by Illumina as a non-indexed library and as such should never appear in demultiplexed Fastq files. In our default analysis pipeline it should only appear in the “lost-reads” file and should be around 1% in data from lanes 1-7, and 5% in data from lane 8 of an Illumina flowcell (the actual percentage of PhiX can vary for several reasons, so we’re not surprised to see higher or lower percentages than expected). We are still running PhiX in almost every lane of sequencing as an easy control to monitor run quality. But if PhiX is getting a barcode what’s going wrong?
The main concern is that if the barcode read is failing in some manner, and attributing barcodes incorrectly, this will lead to erroneous results. There are two major things that index mis-assignment causes
- reads are lost because a spurious barcode was assigned; this data would usually be discarded, should be minimal, and can potentially be ignored.
- barcodes are mis-assigned to the wrong sample; this is a much more serious issue, and understanding what causes it, and the likelihood of it happening, will be critical in reducing the technical factors that could limit low variant calling.
With PhiX on every lane we should be able to monitor index mis-assignment in every run. PhiX may also allow us to estimate the rate of mis-assignment between samples, which will be vital if users need to allow for this in their analysis, particularly in low-frequency variant calling.
Previous reports about multiplexing on Illumina sequencers: As was anticipated several years ago multiplex sequencing has become a common tool in many studies, the level of multiplexing varies but it is almost ubiquitous – an anomaly to this is the creation of indexed libraries in the Genomics England sequencing program but the running of non-indexed sequencing and single-sample-per-lane by the sequencing contractor Illumina.
The Kircher paper presents data from three slightly different preps no-CAP (standard library prep), SP-CAP (single-plex in-solution capture libraries), and MP-CAP (multi-plex in-solution capture libraries). They were able to determine the fraction of mis-tagging events caused by either barcode contamination during oligo synthesis, pooling or handling, by mixed clusters, or by PCR recombination. After removing possible contamination as a source of error they reported that both no-CAP and SP-CAP had low levels of index mis-assignment (0.018% and 0.034%) but that the MP-CAP libraries had more than ten times higher mis-assignment (0.390%). The low percentages in the first tow libraries were due to mixed cluster that could not be eliminated by quality filtering. The high, almost 0.5%, mis-assignment in the MP-CAP library was due to PCR recombination during multiplex PCR after in-solution capture. Importantly they calculated that if this recombination is occurring primarily in the adapter sequences then half of the chimeric reads, almost 0.25% of all exome reads, would be mis-assigned to a sample if a single index was used, and that dual-indexing would be recommended.
Their analysis was confirmed by
Mitra et al 2015 who went further in showing that the template read on the HiSeq was part of the problem – on HiSeq 2500 this is kept to 4 cycles to reduce memory requirements, but when Mitra et al increased template read lengths to 20 cycles they saw 2-5 fold better results for index mis-assignment. Such a long template read would kill most of our HiSeq instruments, but upgrading the memory is suggested by the authors and could be very economical given the impact of low quality cluster detection and index mis-assignment.
In
Jeff’s PhD he used reads from the shortest library molecules with read-through into the adapters to determine that the PhiX control use the older ‘PE’ primers, which have no sequence complementarity to the standard indexing read primers; as such they cannot generate a signal during the index read. He noticed the same drop in quality scores for PhiX index reads compared to the indexed samples as we had. But he also shows that the PhiX reads that appear to be indexed are physically closer to an indexed cluster than PhiX reads with no index read. This led him to propose the same the model of index bleeding as I have here.
Jeff also carefully investigated PCR-mediated recombination (as did Kircher et al) as an additional source of index mis-assignment. This was first reported back at the start of the 1990’s by
Meyerhans et al. In any PCR the polymerase can stall or fall off the template creating a short extension products, this can then hybridise in place of a primer in the next round of PCR. The issue with Illumina libraries is that such a product could create a chimeric index mis-assignment due to molecular swapping of indexes. This is likely to be most pronounced in multiplexed amplification after indexed library prep i.e. most exome and amplicome strategies. He also stated that his analysis “constituted overwhelming evidence that PCR-mediated recombination happens during cluster generation”. His analysis was all on HiSeq 2500 “Manteia” clustering chemistry, this is likely to perform quite differently from the patterned flowcell
“Exclusion Amplification” chemistry and we’re looking into index mis-assignment on that right now.
In the SASI-seq paper Quail et al highlighted the issue of index mis-assignment and discussed the need for confirmation that contamination is not present before a data set is analysed. They presented a simple and inexpensive method to verify that results are not contaminated. They prepared a mix of three uniquely barcoded amplicons, of different sizes spanning the range of insert sizes one would normally use for Illumina sequencing, and added these to samples at a spike-in level of approximately 0.1%. They also designed a set of 384 11bp Illumina indexes sequences with high Hamming distance (5bp apart) higher levels of error correction and very low levels of barcode mis-assignment due to sequencing errors.
Our PhiX mis-assignment analysis results: We took historical data to verify if PhiX mis-assignment was happening across all flowcells and could clearly see this was the case, (A) simply shows the percentage of PhiX we added to each lane. In (B) you can see that the majority of lanes show a reasonably low level of index mis-assignment to PhiX, at just 0.01-1% in single indexed samples (green), and 0.01-0.0001% in dual-indexed samples (red). Dual indexing appears to help significantly. We also saw that the level of PhiX contamination was worse on 2500 than 4000, and increased as the amount of PhiX used increased. In fact the rate of PhiX index mis-assignment was more strongly correlated with the amount of phiX in lane for single indexed samples than for dual indexed samples (C). We see PhiX appearing at as much as 1% of the sample in the very worst cases – however this is generally in single-indexed multiplexed sequencing with very high levels of PhiX e.g. low-diversity spiking.

Indexed versus non-indexed PhiX analysis: Whilst the Illumina PhiX control is not indexed, it is possible to purchase an indexed version from SEQMATIC. When we compare indexed versus non-indexed PhiX the results were clear – non-indexed PhiX shows around 0.02% bleed through, while the SEQMATIC index is around 0.005%; a four fold reduction in bleed through.
 |
| Indexed versus non-indexed PhiX comparison |
Index-read base-quality scores are worthless: We saw that mis-assigned PhiX (PhiX FQ below) reads generally had lower sequence read quality scores than the correctly assigned samples (D). The mis-assigned PhiX index reads were also had generally lower quality scores than the correctly assigned samples (E & F), and it would be great to filter on base quality scores to remove mis-assigned reads. Unfortunately the quality score you get from an Illumina index read is pretty much useless. This is primarily due to its short length. Actually getting the index quality scores requires quite a bit of messing around with the default bcl-fastq pipeline.
These index Q-scores are currently discarded. Just to get the data for the plots below we had to rerun the flowcell through a modified bcl-fastq pipeline. Keeping index Q-scores would require changes to our default pipelines and increase in our compute storage requirements. However we may be able to develop methods similar to Q-score binning, to reduce this extra data, and still allow an assessment of index quality.

Going further than this Illumina sequencing might benefit from running a longer template read at the beginning of all reads e.g. read 1, i5, i7 and read 2. What the computational burden might be and exactly the impact on index mis-assignment this would have is difficult to predict. But even small reductions in errors like this would be worthwhile for low allele frequency applications. I’d expect that companies aiming for tumour screening in the general population (e.g. Grail) would benefit the most from doing these experiments.
PhiX mis-assignment analysis conclusions: Based on our analysis, and the results presented in Jeff’s PhD we’ve come to the conclusion that PhIX index mis-assignment is caused by two issues: index bleeding and/or poly-clonal clusters. And that this can be fixed or safely ignored.

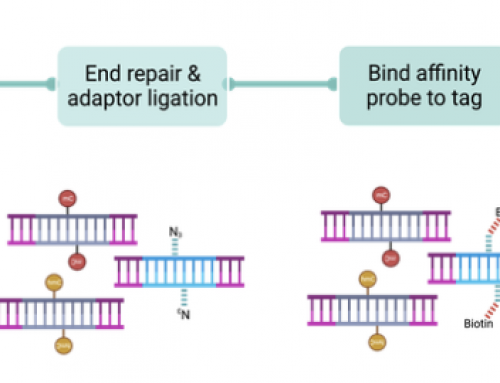
In the figure above (1A) I’ve tried to present “index bleeding†– each library template cluster emits a signal according to it’s base-fluorophore, represented by the capitalised circles as GAT, (green=G/T, red=A/C), however this fluorescent signal “bleeds†outward from each cluster. A non-indexed PhiX cluster, represented by the lower-case circles, does not emit signal and is base-called from the erroneous “index bleeding” library cluster signal as gat. An indexed PhiX cluster emits a signal according to it’s base-fluorophore and is correctly base-called as CTA. In figure 1B I’ve tried to present what may be happening on mixed template poly-clonal clusters. These are caused by the random nature of clustering where some clusters are made from two template molecules, that may have seeded at different times. A cluster produced from a single library molecule (α) is correctly base-called as GAT. A mixed template non-indexed PhiX cluster (β) is base-called on the low-signal from the erroneous library cluster signal in the indexing read only, due to lack of PhiX index signal as gat. A mixed template indexed PhiX cluster(γ) emits a signal according to it’s base-fluorophore that is higher than signal from the erroneous library cluster and is correctly base-called as CTA.
Index-bleeding should only be an issue for non-patterned flowcells, whilst poly-clonal clusters will be a problem on both patterned and non-patterned flowcells i.e. HiSeq 4000 and 2500.
How to fix the problem: for index mis-assignment to PhiX the fix is relatively straight-forward. Either use an indexed PhiX, or spike in an oligo to the indexing read primers such that PhiX generates a signal. Both strategies will mean the PhiX clusters generate a signal that outcompetes the index-bleeding, or poly-clonal cluster signals. PhiX will no longer appear in your demultiplexed fastq, or will be at such low levels you’d only see it if you specifically went looking.
Unfortunately index mis-assignment between samples is still an unresolved issue. In a follow up post I’m going to discuss what we’ve seen, and what the apparent causes are. Again some relatively simple fixes are available – but if you are using multiplexed sequencing to detect low-frequency alleles in populations; e.g. cancer, single-cells, population genomics, then you need to consider whether you understand how your experiments might be affected.
PS: I think it is pretty lax of Illumina not to provide an indexed PhiX. The V2 PhiX was indexed but V3 dropped this, probably due to there only being 96 TruSeq indexes. Come on Illumina sort this one out!












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
We see Index bleed quite frequently on the HiSeq 2500. Especially when the multiplex balance is not sufficient to provide representation of all 4 bases in each position of the index read. I can also concur that this problem increases as the PhiX spike-in increases. We have low-diversity samples that need an increased %PhiX to overcome difficulties in the first 20 bases. If we don't have at least 12 samples in the pool, with every base represented in the entire index read, the data is virtually useless and cannot be demultiplexed. In addition to this problem, we have also noticed that with an under-representation of A's and C's in the index, the software is likely to call N's as A's and C's incorrectly. Why these bases, we don't know, but it seems to be just those two.
The two-color chemistry on the NextSeq (and presumably the MiniSeq) is a whole other problem, because a cluster with no signal is going to look the same as GGGGGG. Which is also only two bases off from TruSeq LT index 23 (GAGTGG), making that index unusable with two-color chemistry. It makes me worry a lot about sample-to-sample bleeding where the difference between index sequences depends on G too.
We noticed that samples mis-assigned to the wrong index are coupled with generally poor index quality.
https://www.ncbi.nlm.nih.gov/pubmed/27183494
The upshot is the introduction of a custom bcl2fastq replacement that discards reads accomanied with poorer index seqeunce.
https://github.com/edm1/error-aware-demultiplexer
Hi Unknown (Jack or Mike?),
I saw the paper but cannot access it as it is behind the JMD firewall 🙁
We also noticed that bad indexes appear to be coupled to lower quality. However we got a very string steer from Illumina that the length of the index reads made any assessment of their quality pointless. If you can do better than them then I’d like to know more. Can you send me a copy of the paper? Have you considered doing longer index reads to improve their quality assessment?
James
Hi,
Have just recently stumbled on to this page… great resoure James….
We had also observed that miss-assignments are related to the index quality. However, in a related note we have noticed that allowing 1 mismatch during demultiplexing contributes very highly to the miss-assignments, so usually we don’t allow mismatches (on top of applying a quality filter to the index). The thing is that with runs have a problematic index run one is tempted to use mismatches to recover yield, however those situations are exactly the ones prone to miss-assignments. So overall when the run is good the mismatch has a minor effect, when the run has index problems one could be promoting miss-assignments; so overall I think its best not to use the mismatch option.
RA
We also tend to use 0 mismatches. Illumina’s index reads are really not long enough to allow quality filtering to be applied. If there is any suspicion of a poor index read contributing to the failure of a run to generate enough data we’d always push back on Illumina to replace the entire run.
Hi,
I found this post highly informative (though I must admit, being a newbie on the field of NGS, it is portion that I could actually fully understand). But I would place my question here, you guys might help me out with a problem that is just the other way around than yours (you having PhiX inflitrate your demultiplexed samples, while it is me having most of my reads ending up in Undetermined reads folder). Just in nutshel: I run my first RNAseq library (12-plex, Scer poly-A targeted cDNA) on a Miseq with 5% PhiX (unindexed) spike in. Two problems occured: the run was severely underclustered (Problem1) and those few reads (3.5 million) that were generated mostly ended up in Undetermined folder. Mapping content of the undetermined folder gave 37% of it mapping to our target genome (Scer) and 50% mapping to PhiX genome. Demultiplexed samples were also lousy regarding index quality: 70-80% indexes had 1 mismatch (Demultiplexing was done automatically by Miseq, I am not using yet any specialised demux tool). What do you think the problem was? I suspect it is somewhere around index sequencing quality. How could I improve the run? I will increase library concentration and thinking about decreasing PhiX%.(And a very basic bonus question: where exactly reads from the unindexed PhiX are supposed to end up? I somehow get the idea that in Undetermined folder, only reads that have some kind of index sequence read should turn up. Then how come I see PhiX reads in Undetermined folder? Is it index bleeding?) Well, thank you so much, I hope someone gets my message…:) Greets!
Sorry Krisztina, I’m not a bioinformatics expert so ca’t really help. I’d suggest you jump over to SEQanswers or BIOstars