 |
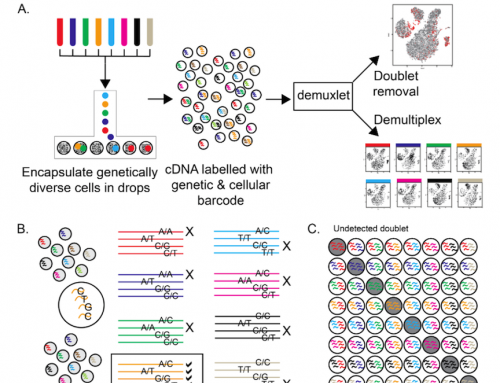
| Adapted from Figure 1 in Hicks et al. |
Almost without exception every new technology gets published with a slew of high-impact papers. And almost without exception those papers turn out to be heavily biased. This is not to say we should expect every wrinkle to be ironed out before initial publication – new technologies take a lot of effort and the faster they make it into the public domain the sooner the community can improve them and make them more robust. Often batch effect is the first problem identified: with arrays, with NGS, and now with single-cell RNA-seq.
Stephanie et al looked at 15 published single-cell RNA-seq papers and found that in the 8 studies investigating differences between group, and where they could assess confounding effect it ranged from 82.1% to 100% (see table 1 from the paper – 82,85,93,96,98,100 & 100%). All of these studies were designed such that the samples were confounded with processing batch. They report that the number of genes detected expressed explained a significant proportion of observed variability, but that this varied across experimental batches. This confounding of biological question with experimental batch effectively cripples the project;
However the authors do point out that relatively simple experimental design choices can be used to remove the problem.
- Batch effects can be a big problem in scRNA-Seq data (but not always).
- Batch effects and methods to correct for batch effects have been around for many years (lots of places to start).
- Bad news: Poor experimental design is a big liming factor…. also, more complicated because of sparsity (biology and technology), capture efficiency, etc
- Good news: Increase awareness about good experimental design. New methods specific for scRNA-Seq are being developed
I’m putting effort into understanding spikes in a lot more detail and am sure we’ll all be using them routinely in a few more months.
What does this mean for the choice of scRNA-seq platform: My briefest of surveys for the three platforms we’re using or looking at in my lab are as follows. Fluidigm suggest using the ArrayControl RNA Spikes (Thermo Fisher Scientific AM1780). Drop-seq suggest using the ERCC spikes (although this is not mentioned in their online protocol). 10X Genomics don’t say anything about spikes in their current protocols!
I generated the figure at the top of this post to show where these 3 scRNA-seq platforms fit into Stephanie’s figure 1 from the paper. Both C1 and Drop-seq are completely confounded as only one sample is processed per batch. 10X Genomics allows up to 8 samples to be processed together so a replicated “AvsB” study could be completed with zero batch effect.
But in the future we’re likely to need 12, 24 or even 96 sample systems that allow us to process a scRNA-seq experiment in one go. Whilst it may well be possible to design Fluidigm C1 chips that can process more samples, each with fewer cells, or for Drop-seq to emulate 10X Genomics, or even for 10X Genomics to move to a larger sample format chip; none of this will solve the problem of collecting large numbers of single-cell samples without introducing batch effects further upstream in the experiment.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave A Comment