I’m continuing my exome posts with a quick observation. There have been several talks recently that I’ve seen where people present genome and exome data and highlight the drop-out of genomic regions primarily due to PCR amplification and hybridisation artefacts. They make a compelling case for avoiding PCR when possible, and for sequencing a genome to get the very best quality exome.
A flaw with this is that we often want to sequence an exome not simply to reduce the costs of sequencing, but more importantly to increase the coverage to a level that would not be economical for a genome, even on an X Ten! For studies of heterogeneous cancer we may want to sequence the exome to 100x or even 1000x coverage to look for rare mutant alleles. Unfortunately this is exactly the kind of analysis that might be messed up by those same PCR artefact’s, namely PCR duplication (introducing allele bias) and base misincorporation (introducing artifactual variants).
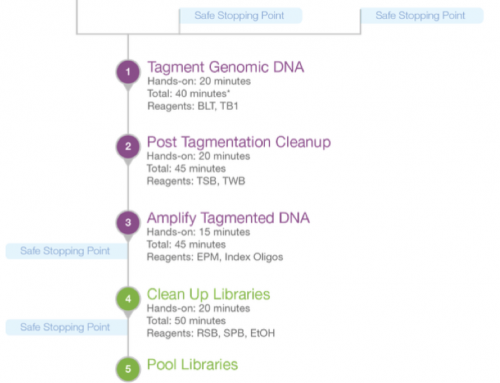
PCR free exomes: In my lab we are running Illumina’s rapid exomes so PCR is a requirement to complete the Nextera library prep. But if we were to use another method then in theory PCR-free exomes would be possible. Even if we stick to Nextera (or Agilent QXT) then we could aim for very low-cycle PCR libraries. The amount of exome library we are getting is huge, often 100’s of nanomoles, when we only need picomoles for sequencing.
Something we might try testing is a PCR-free or PCR-lite (pardon the American spelling) exome to see if we can reduce exome artefacts and improve variant calling. If anyone else is doing this please let me know how you are getting along and how far we can push this.





{kind=link}
{kind=link}
Dear James,
How do you think will be difference between PCR and hybridisation approaches in case when we don't search somatic mutations?
Hi Anon,
If you are looking for heterozygous variants at normal Mendelian frequency then I'd guess PCR is not such a porblem. There were reports of reference allele bias early on but I think this is less of a problem today. If you have plenty of DNA I'd still go with PCR free methods if I could but we need to see these being made available with exome capture kits.
Thank you very much for answer