Carlos Caldas, Functional genomics of cancer: perturbation experiments in the lab and in the clinic. Carlos is a senior group leader at the Cancer Research UK Cambridge Research Institute where I work, he is also a practising oncologist and one of the founders of CRI. Carlos stood in at short notice for Eric Lander who had to stay behind to advise Barack Obama, during the upcoming election!
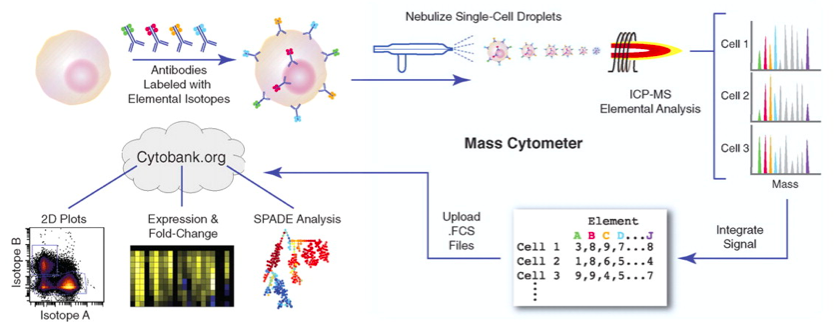 |
| Mass-cytometry image from Bendall et al: Science 2011 |
IAP antagnoisists, inhibitors of apoptosis by binding to caspases, SMAC can repress this inhibition by binding to the IAP proteins. At Novartis they have been searching for XIAP agonists LCL161 is teh fruit of their search. There are 7 or8 bona-fide IAP family members, ideally we would knock out all of them. Wehn testing LCL161 it became clear that although designed to one IAP they actually inactivate another family member at much higher efficiencies, and that this family member does not bind caspases! The drug appears to work by inducing TNF driven apoptosis. They are aiming to kill cancer cells rather than just stop growth.
Only 7% of cell lines are sensitive to LCL161, is it possible to predict which cell lines or even patients will respond? They are using data and cell lines generated for the cancer cell line encyclopedia and presented a gene expression signature for response to LCL161. It is not jst gene expression being run, they are now collecting CNV, methylation, exome-seq etc across 1000 cell lines treated with multiple parameters.
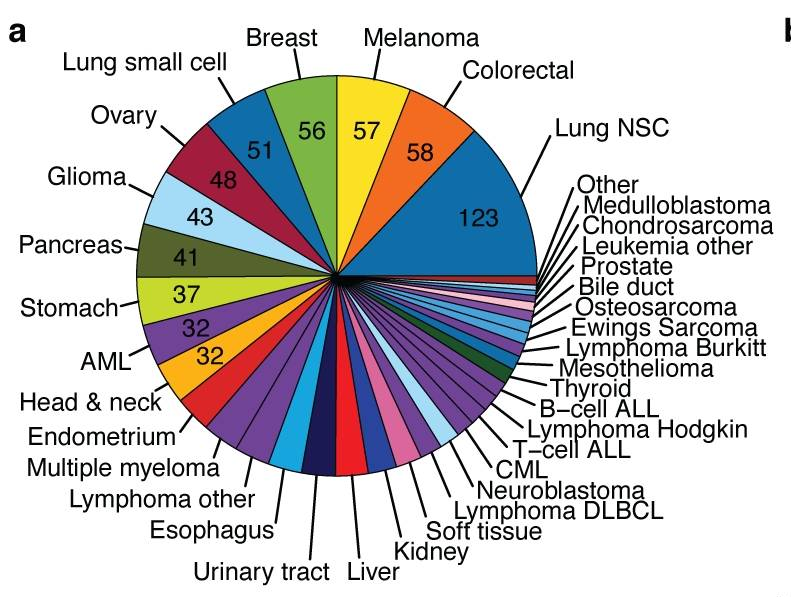 |
| cancer cell line encyclopedia cell lines |
Cell line encyclopedia was used to look for compounds that might target triple negative breast cancers. Three candidates were found and all were IAP inhibitors. Digging in the data using pooled shRNA analysis of 250 cell lines using representation sequencing after selection shows a novel functional genomics screen that should help in target discovery. They asked if any shRNAs were depleted in triple negative breast cancers vs other breast cancer cell lines? Again XIAP targeting shRNA came up as targets.
Rebecca McIntyre, High-throughput functional validation of candidate colorectal cancer genes. Rebecca is a PhD student in David Adams research group at the Wellcome Trust Sanger Institute.
Rebecca presented work on validation of three colorectal cancer genes. Colon crypts are very well studied and complex organs. Hyperactivation of WNT pathway is often seen in CRC. The 1990’s brought us the canonical APC, Kras, Smad4, TP53 Fearon and Vogelstein model. More recent work in Science 2006 & 2007 identified novel genes and four fo these were selected for validation in APCmin mouse models.
MLL3: a chromatin modifier which was the fourth most mutated CRC gene, it is a large gene with multiple polyA regions both of these make it more likely to be mutated. Is it a bona-fide CRC driver gene? ChIP-seq analysis shows that MLL3 down regulates AXIN2 expression which is involved in WNT signalling. Crypts of MLL3 deficient mice are longer and may also explain why tumouregensis is higher. Rebecca also presented work on PKDH1 and PARK2.
Jason Carroll, Understanding oestrogen receptor transcription in breast cancer. Jason is a senior group leader at the Cancer Research UK Cambridge Research Institute where I work.
- Where does ER bind to DNA
- How does it bind to DNA
- Are there similarities or differences in ER binding in a resistant or sensitive context?
Jason’s guess is that there are probably 50,000 DNA binding sites for ER. His previous work was instrumental in the discovery of FOXA1 as a “pioneer factor”. More recent work in other labs is allowing study of distant binding sites through chromatin loops.
Nearly all work in ER is being performed in MCF7 and of course this can not possibly represent all ER biology. Work in Jason’s lab also uses primary breast cancer tumours and ER ChIP-seq showed clear classification of samples as good, poor or metastatic. Differential binding was seen in good vs poor vs metastatic samples. For three tumours they split the sample in two to get some idea of the impact of tumour heterogeneity, replicates gave highly concordant data. They looked to see if the differential binding switched on different genes but did not have RNA for analysis. They developed a gene predictor and tested it in multiple cohorts where it was shown to correlate with outcome, but only in ER+ve samples.
ERE and Forkhead motifs turn up time and again in poor outcome samples. Data from three different ER+ve cell lines show that generally when FOXA1 moves in the genome ER binding follows. Differential binding also occurs in tamoxifen resistant cell lines when compared to susceptible
ER binding can be reprogrammed in a rapid timescale. Growth factors EGF and IGF can induce tamixofen resistance in just five minutes by reprogramming ER binding in the genome. But what happens to FOXA1? It turns out that mitogen induced ER binding correlates with FOXA1, so what allows FOXA1 to bind?
How do you find the pioneer factors pioneer factor? Using a modified Chromatin IP and proteomic analysis of the total bound protein. They specified that three replicates had to show the same results and that IG controls must be silent. They found 108 ER protein:protein interactions and can do this in patient samples. For FOXA1 they found 24 proteins, many of which were chromatin modifiers and hormone independent preparing FOXA1 for potential ER binding. Are there post-translational modifications of FOXA1? Are there differences in a resistance context?
How important is FOXA1 for ER function? It is essential, if FOXA1 is silenced ER signal drops by 90% even though the protein is still present. Naked DNA oligos will still bind ER as it is not chromatinised. A word of caution for reporter assay interpretation. FOXA1 seems to be the sole determinant of ER function and is one of the signature genes of ER+ve breast cancer (HNF3alpha aka FOXA1). They are now collaborating with Carlos on larger numbers of patient samples.
The big question is whether FOXA1 is going to be a suitable drug target?
“Unanswered questions” panel discussion:
We have the technology to ask big questions about functional genomics of cancer and bring this to the clinic. So how are we going to do this when the scientists do the research, the clinicians have the patients and the drug companies have the drugs?
How will sequencing impact our choices of drug in the clinic?
Can we determine up front which combinations to give at the start of treatment?
Can we better understand which patients should not be treated?









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave A Comment