Go back to the summary of the CRI symposium.
Barbara Wold, The genomics discovery path from TCGA and TARGET to the clinic…and back. Unfortunately Dr Wold flew all the way over, escaping hurricane Sandy but was kept in her hotel room by flu.
Keith Peters, was Regius Professor of Physic at the University of Cambridge from 1987 to 2005, where he was also head of the School of Clinical Medicine. Under his leadership the University’s Clinical School became a major centre for medical research, complementing Cambridge’s strengths in basic biomedical science.
Sir Peters last-minute presentation started with a little look back to 11th April 1988 and a photograph of a document he had written with Sydney Brenner for a meeting with Prime Minister Margaret Thatcher. Sydney Brenner and Keith Peters were “selling” the UK involvement in the Human Genome Project as at the time the MRC was not particularly interested. there were lots of scribbles from Keith and Sydney debating on what the impact would be of the final project on medicine.
Lori Friedman, Overcoming resistance to targeted therapies in breast cancer. A former post-doc with Bruce Ponder and Mary Claire-King now working at Genentech where she is senior director of translational oncology.
Dr Frieman presented two themes in her talk, how mutations can affect choice of therapy, 2nd mechanisms of resistance, innate and acquired and how evolution impact this. Questions; the issue of over-treatment, heterogeneity, what to do with infrequent mutations, druggability of loss-of-function mutations.
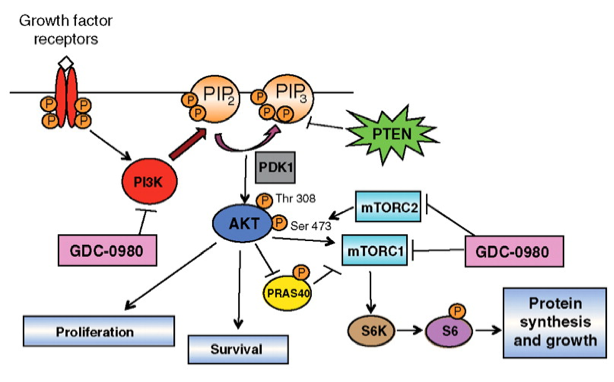
She used PI3Kinase as an example; it is the most frequently altered pathway in cancer and PIK3CA is the most commonly mutated druggable oncogene where mutations were identified in 2004 (
Samuel’s et al Science 2004) and have been shown to increase kinase activity and activate downstream signalling. There are over 20 PIK3CA inhibitors in over 150 clinical trials, a massive investment in a single druggable target!
GDC-0941 is the Genentech PIK3CA inhibitor in clinical trials. Using differential gene expression to determine which samples might be responsive to the drug, 17 gene signature. Suggestive of a feedback mechanism where cells may reset their sensitivity to activation of pathways and become resistant. Clinical protocol starts low and goes higher as tumour cells adapt. When cells get to 10x EC50 single cell clones are selected for analysis to determine mechanisms of resistance. Sequence PIK3CA, GX, CNV, RPPA? and genome sequence. Combination therapy with dual PI3K/mTOR inhibitors rescues resistance to the GDC treatment. Analysis of clones shows PTEN loss and resultant increase of pAKT.
 |
| Cell pathway and mechanistic effects of GDC-0980 treatment in tumor cell lines |
Peter Lichter, Sequencing of paediatric brain tumours: From molecular profiles to clinical translation. Division of Molecular Genetics, at the German Cancer Research Centre.
Pediatric Brain tumours are the most common cause of mortality in children. Dr Lichter spoke about three different paediatric brain tumours and sthe work ongoing at DKFZ as part of the ICGC. Medulloblastoma stratification currently based on age, metastases and degree or resectability, recent genomic works shows consensus (
Taylor et al 2012 Acta Neuropathalogia) of four subgroups. Dr Lichter’s group wanted to extend the detail of this work. So far they have shown a background mutation rate that is quite low, only around 10 non-synonymous coding mutations and 588 of 765 SNVs were private to an individual (
Jones & Jager 2012 Nature). Another word cloud with just a few big words, the cloud was very nicely coloured by subgroup (how did they do that?). There is a correlation between the age of the patient and the number of mutations, not seen in other tumours, what is the mechanism behind this? Their data is suggestive of different subgroups having different cell-of-origin, also global hypomethylation patterns show subgroup specific profiles.
 |
| Peter Lichter’s word cloud coloured by stratification group |
Marlous Hoogstraat, moving towards implementation of next-gen into clinical decision making. PhD student at UMC Utrecht, centre for personalised cancer treatment and the research school Cancer Genomics & Developmental Biology.
Marlous presented a collaboration between three centres NKI, UMC Utrecht and Erasmus Medical Center. She described their sequencing approach where they are allocating patients with metastatic disease into clinical trials, taking 2-4 matched biopsies, performing pathological analysis, DNA isolation and then completing a patient stratification using Amplicon sequencing on Ion Torrent and MiSeq. They try to allocate patients to a trial and monitor resistance or progression, taking repeated biopsies to understand results. All in two weeks! They are running 5500 and starting up wildfire to sequence 2000 genes and are moving to whole exome.
The rate limiting step for the analysis is not the sequencing. The logistics are a hurdle, collection and transport of samples, pathological review, batching for processing and sequencing all add time, whilst the sequencing itself is completed in a day or two.
Biopsy protocol: perform image guided biopsy, pathologist selects tumour rich areas for DNA extraction. All tissue is snap frozen. DNA yield and tumour percentage across the first 100 samples showed generally more than 1.5ug of DNA and most tumours are 50% cancer cells although they are using samples with just 20% tumour in very high depth targeted sequencing. Presented results from Sunitinib trial, TP53, KRAS, APC, KDR, Met, CTNNB1, FLT3, PTEN, EGFR, FGFR3, SMAD4, IDH1. They found different numbers of mutations per patient and 11 that have no mutations (silent genomic signature?), there were no strong associations with aggressive or indolent disease. Now moving to Stratification 2.0, including complete tumour suppressors and amplifications.
Data being reported back after multi-disciplinary team meeting (how much do these meetings cost?) The 2000 genes being sequenced in their research track analysis are being captured using a custom Agilent SureSelect product.
Nitzan Rosenfeld, Circulating tumour DNA as a non-invasive tool for cancer diagnostics and research. Nitzan is a group leader at the Cancer Research UK Cambridge Research Institute where I work.
He presented his groups work on ctDNA mutational analysis. Cancer is an evolving disease of the genome and ctDNA provides a way to monitor this in real-time. A problem is the ctDNA contains normal DNA potentially in a high background. His talk started with an experiment where a tumour was initially sequenced for personalised genomic biomarkers and then ctDNA was analysed for these markers in a follow-up study. Comparing ctDNA to CA125 as a marker for high-grade serous ovarian cancer and demonstrating that ctDNA can be informative when CA125 fails. The patients were analysed with digital PCR over 600 and 300 days during which they were treated with secondary and tertiary therapies as tumours relapsed. ctDNA gave a faster sharper response with greater significance in a prognostic setting. The response is probably even sharper but the study is limited by the sampling of blood being 3 weeks after therapy.
His group is also collaborating with Carlos Caldas and Sarah-Jane Dawson. And he presented results that suggest in metastatic breast cancer patients rising ctDNA levels often predate radiological disease progression or a change in CTCs or CA15-3 levels.
He also spoke about a third collaboration showing how it is also possible to look for mutations that could be used in targeted therapy. The collaboration with Tan Min Chen Singapore, looks at acquired resistance to anti-EGFR therapies.
Targeted amplicon sequencing
TAm-seq is a practical compromise that allows a sizeable and useful portion of the genome to be sequenced with similar sensitivity to single-locus assays. And it is dirt cheap! How much can we get from ctDNA, only time wil tell?
Bruce Ponder, How can we use knowledge of genetic variation? Bruce is Director of the Cancer Research UK Cambridge Research Institute where I work.
Inherited cancer predisposition is becoming more and more a computational problem but it is important to understand the clinical challenges as well. Cancer susceptibility is well understood in a familial BRCA1/2 context, but only counts for about 15-20% of risk and under 5% of Breast cancers. Missing heritability is buried in the normal genetic variation. In 2000 there were 12 predisposition loci, today over 1000 in 165 different diseases. Consider the predisposition genotypes as a hand of cards dealt at birth, your hand will put you somewhere on the normal distribution. Understanding the mechanism of action of the individual loci will help us better understand cancer, ideally we can understand risk as well and target therapy better.
FGFR2 per allele risk is low at just 1.26% of GWAS heritability, what does this mean in the clinic? Normal lifetime Breast cancer risk is 9.7%, a single copy of the FGFR2 variant increases this to 10.2% and two copies is still just 13.4%. This is not huge.
Introducing “attributable fractionâ€: Prof Ponder gave the example of Breast cancer where the attributable fraction of being a women is 99%, so if we got rid of women we would reduce the number of breast cancers massively. Obviously this is not a practical solution. FGFR2 has a significant attributable risk and if reduced to normal would eliminate 16% of breast cancer. Great for public health but what are the effects of taking FGFR2 inhibitors over a long term? But there are good approaches in other diseases, e.g. statins that target the activity of a single enzyme to lower cholesterol and reduce heart disease.
“Unanswered questions” panel discussion introduced by Kieth Peters
Is prediction of individual risk by SNPs currently clinically useful?
How will we do clinical trials on what are quickly becoming orphan diseases due to stratification?
How can we target tumour suppressors or other undruggable targets? Is synthetic lethality.
How do you get the dose right in a combination therapy?
How do you generate a combination therapy when your competition has the combinatorial?
Is drug development “self-harming” in that drug companies are all chasing the same target where we will ultimately need combination therapies to reduce the rate of resistance so those companies should work harder on different targets and collaborate on combinations?
Like this:
Like Loading...
Related










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave A Comment