This post follows on from my previous one explaining the 10X Genomics single-cell mRNA-seq assay. This time round I’m really reviewing the method as described in a paper recently put up on the BioRxiv by 10X’s Deanna Church and David Jaffe: Direct determination of diploid genome sequences. This follows on from the earlier Nat Methods paper which was the first 10X de novo assembly of NA12878, but on the GemCode system. While we are starting some phasing projects on our 10X Chromium box the more significant interest has been on the single cell applications. But if we can combine the two methods (or something else) to get single-cell CNV then 10X are onto a winner!
The paper describes the 10X Genomics Chromium phasing technology. They highlight the impact of their tech by first reminding us that the majority of Human genomes sequenced to date are analysed by alignment to the reference (an important point often forgotten by users). They say that only a few de novo Human assemblies have been created, but that most do not truly represent complex biological genomes. The authors only consider two published genomes as true diploid de novo assemblies – Levy et al. PLoS Biol 2008: The diploid genome sequence of an individual human and Cao et al. Nat Biotech 2015. De novo assembly of a haplotypeÂresolved human genome.
The method: They introduce the 10X Chromium library prep. This starts with 1.25ng of >50kb DNA, from which 16bp barcoded random genomic loci are copied (by polymerase extension?) inside the Chromium gel-beads. Each of these contains around 10 molecules per droplet equal to ~0.5 Mb of the genome. The most important bit of the tech is the ability to put just 0.01% of the diploid Human genome into a single droplet – this makes the probability of both alleles being present vanishingly small. With 2 lanes of X Ten you can expect to get about 60X Human genome coverage and the authors calculate the number of “linked reads” per molecule as 60, which equates to around 0.4x coverage (enough for shallow CNV sequencing to reveal clonality in Tumours perhaps).
Question to the authors: I do not understand the statement about smaller genomes getting lower linked read coverage: “For smaller genomes, assuming that the same DNA mass was loaded and that the library was sequenced to the same readÂdepth, the number of LinkedÂReads (read pairs) per molecule would drop proportionally, which would reduce the power of the data type. For example, for a genome whose size is 1/10th the size of the human genome (320 Mb), the mean number of LinkedÂReads per molecule would be about 6, and the distance between LinkedÂReads would be about 8 kb, making it hard to anchor barcodes to short initial contigs.” My first assumption was that genome size would have no impact on linked read depth, but it would significantly affect the amount of the genome present in a single droplet. As such the smaller genome, with DNA fragments of the same size should still have around 60 linked reads per DNA molecule, but a 10MB genome would mean 5% was in each droplet making the phasing much harder to determine. Please feel free to explain this to me.
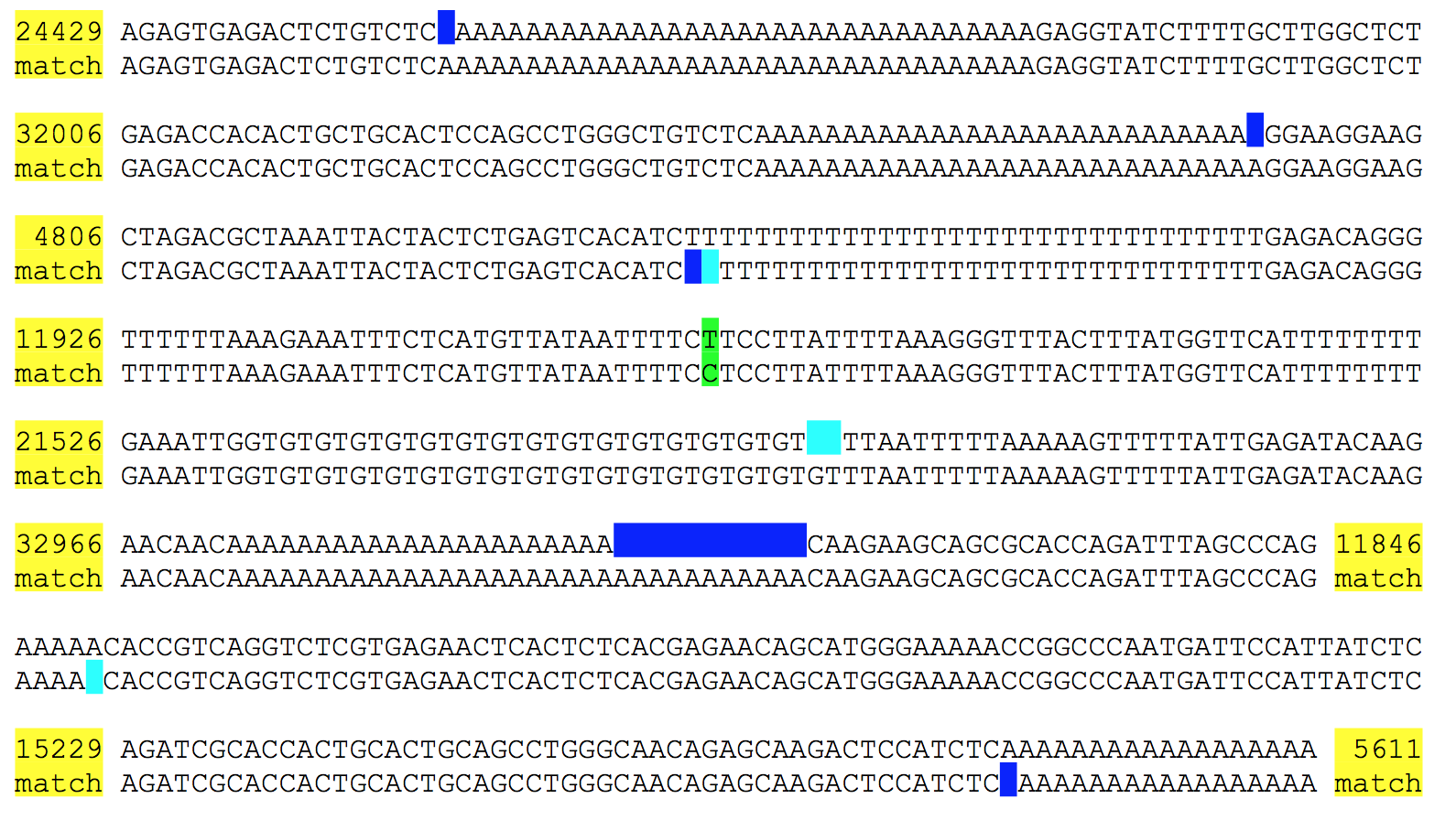
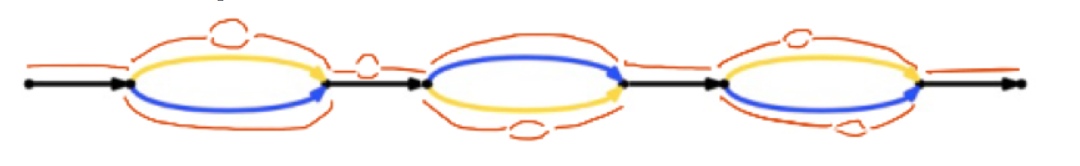
The data: In the paper they present data from seven Human genomes, sequenced on HiSeq X Ten, and assembled using the “pushbutton” Supernova algorithm (it won’t run on your Mac Book Pro as you’ll need >384Gb of RAM). In just two days per genome they generated 100kb+ contigs with 2.5Mb phase blocks. The 7 genomes include 4 with parental data to verify phasing results, as well as one sample used in the HGP. They include a figure (see below) showing the Supernova assembly of the HGP sample aligned to a 162kb clone which is part of the GRCh37 reference. It almost completely matches the reference sequence with the 8 variants including just 1 SNV (green), but 6 homopolymer and 1 di-nucleotide repeat length variants (blue/cyan). The sceond figure shows the representation of the path a FASTA sequence takes through the “megabubbles” separating parental alleles, and “microbubbles” caused by longer repeats and homopolymers.
 |
| Who’s careful hand at 10X Genomics drew this representation of FASTA?
|
Tuning 10X phasing to your needs: Users may be able to “tune” scaffold N50 by varying DNA length or sequencing coverage. A single X Ten lane generating 30x coverage looks like it would push scaffold N50 down from 17 to 12 Mb. DNA quality is probably most important and I suspect many people will accept a significant improvement in phasing estimation from lower cost experiments.
Many groups will also want to run differently sized genomes and will need to estimate how much DNA to use and how much sequencing they’ll require. For small genomes this gets really interesting and 10X could be an awesome metagenomics tool allowing strain level analysis of complex samples. For the larger non-Human genomes people will need to us a much smaller amount of DNA in a single run, which may limit the number of genome copies to an unreasonable level.
- Human 3Gb = 1ng = 300 genome copies
- Wheat 5Gb = 0.67ng = 135 genome copies
- Maize 20Gb = 0.17ng = 8 genome copies
- Salmander 50Gb = 0.07ng = 1.3 genome copies
- Paris japonica 150Gb = 0.02ng = 0.15 genome copies
Who’s going to use Chromium phasing: Is this kind of data going to be relevant enough for people to adopt 10X Chromium as the default genome library prep? I suspect many teams are working on 100s or even 1000s of 10X Genomics genomes right now and we’ll see many more publications very soon. If the $500 Chromium prep can add real value (biologically or clinically) then 10X have a real chance of becoming a new standard for library prep. If that’s the case I guess we’ll see how strong their IP is as the competition builds their own variants of the technology.
Like this:
Like Loading...
Related








{kind=link}
{kind=link}
{kind=link}
{kind=link}
James – curious on your thoughts regarding data quality differences between 10X+Illumina and Pacbio sequencing. Especially the argument that Pacbio generates "contigs" while 10X is "scaffolds" with gaps. Thanks.
Hi Anonymous,
We’ve spent much more effort on the single-cell methods from 10X, and my lab does virtually nothing with respect to genome builds…as such I don’t really think I can comment on whether 10X “scaffolds” or PacBio “contigs” would be better/worse than the other. I’m expecting we’ll see a rush of papers in early 2017 from both technologies as early users demonstrate what’s possible. And I’d be surprised if we did not also see a direct comparison using some of the more difficult genomes out there.
James.
[…] Previous Next […]