The number of 10X Genomics publications is going to grow rapidly; and this list will only be updated sporadically!
 |
This paper by Deanna Church and David Jaffe et al describes the 10X Genomics Chromium phasing technology. I’ve done a more comprehensive write up of this paper here on Core-Genomics. Essentially this is the paper to refer to if you’re considering using Chromium phasing in your own research and want to better understand how it works and what you can do. The authors explain the basic principles of generating LinkedReads, and present data on 7 Human genomes successfully assembled from HiSeq X data using the Supernova algorithm. Assemblies are good with 100kb+ contigs and 2.5Mb phase blocks, and the HGP sample used had excellent alignment to the reference along a 162kb contig.
The authors present ABySS 2.0 and compare it to the previous version and 5 other assemblers, BCALM2, DISCOVAR, Minia, SGA and SOAPdenovo. They used the Genome in a Bottle data: 70X coverage Human genome using Illumina paired 250bp reads (PE250) as well as mate-pair data, 10X genomics Chromium data, and BioNano optical mapping data. ABySS 2.0 generated an N50 of 3.5 Mb using only 35 GB of RAM (still won’t run on your Mac Book Pro). Whilst this is not a 10X paper per se they do discuss the limitations of current short-reads and the impact the 10X technology is likely to have on assembly including the BioNano Genomics and 10x Chromium data increased N50 from 29 to 42 Mb. In Fig. 3 from the paper (see below) the authors show all of the 90 scaffolds over 3 Mb, which add up to 90% of the genome. And state that “most chromosome arms are reconstructed by 1 to 4 large scaffolds”.
 |
| Fig.3 from Jackman/Vandervalk et al 2016 |
The authors present a hybrid assembly of NA19240 using multiple technologies including PacBio, BioNano genomics, Illumina sequencing, 10x Genomics LinkedReads, and BAC hybridization and sequencing. They explain the need for multiple technologies given that no single method “can fully resolve every genomic feature and/or region”; and argue that BAC tiling is still a useful technology. I’d be interested to know how useful this might be once 10X Genomics becomes standardised as the time and cost involved in BAC library construction, mapping and sequencing, let alone the huge amount of DNA required is quite outside the reach of most labs.
The assembly presented is the first in a set of 5 genomes which the authors are aiming to use to improve the diversity of the reference genome. They refer to “Gold” and “Platinum” genomes but I cannot tell which the final assembly was considered. The final assembly had an N50 of 7.25 Mb and a scaffold N50 of 78.6 Mb, which according to the authors “represents one of the most contiguous high-quality human genomes”.
This paper describes a combinatorial approach to de novo assembly and phasing analysis using Illumina sequencing, 10X Genomics (GemCode) LinkedReads, and BioNano Genomics mapping; again using NA12878.
 |
This paper describes the 10X Genomics single-cell 3′ mRNA-seq technology. I’d previously covered this paper here on Core-Genomics. Essentially this is probably the paper to read to if you’d like to are considering 10X Genomics single-cell RNA-seq in your own research and want to better understand how it works and what you can do. The authors explain the basic principles of the methods, and present data from 250,000 cells across 29 samples. An awesome paper…when does it come out in a Jurnal?
Ben Hindson (10X Genomics CSO) will be presenting this work at the San Diego Festival of Genomics if you’d like to know more.
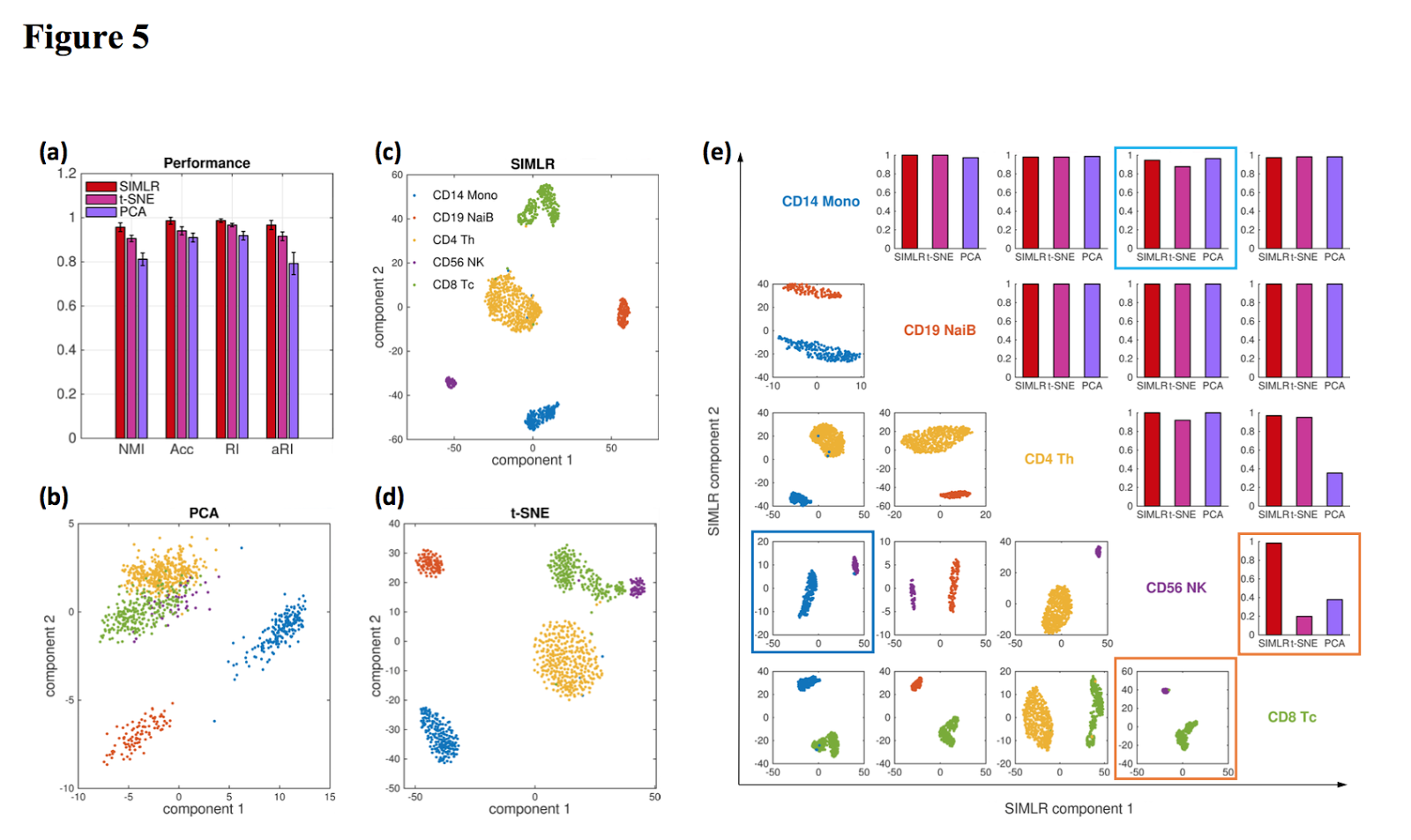 |
| Fig 5: 2D visualisation of data from 5 cell sub-populations by PCA (b) SIMLR (c) and t- SNE (d). |
This review of third-generation NGS systems describes 10X Genomics Chromium genome technology as a mapping, rather than a sequencing application. 10X Genomics is lumped in with BioNano Genomics, Dovetail Genomics cHiCago (HiC) method, genetic maps and mate-pair mapping. The paper includes a great table highlighting the characteristics of the different 3rd-gen platforms (reproduced below).

This paper from Hanlee Ji’s group at Stanford and 10X’s Ben Hindson et al describes the 10X Genomics GemCode phasing technology. It is the first paper to demonstrate that droplet methods for phasing and structural variant analysis. This is the other paper you should refer to if you’d like to are considering phasing in your own research, but the more up-to-date Chromium BioRxiv Church/Jaffe paper (see above) will give you better information about the technical performance today (Sept 2016).
 |
| Fig1: !0X technology overview |
This paper demonstrates what can be done for cancer genomes, and that is what makes it such an important read for people deciding if the 10X the might be useful in their research here at the CRUK Cambridge Institute. I’ve previously written about why I’m excited about using phasing to resolve complex structural rearrangements and determine if multiple variants in the same gene are in cis/trans (cis- is on the same allele, trans- is on the second allele).
For a single colorectal cancer patient they generated 50x Illumina WGS and 30x 10X Genomics WGS (the choice of the name 10X Genomics requires some explanation, written as it is here only the inclusion of the work “genomics” in the sentence makes it easily interpretable. And given that most 10X Genomics phasing data will be generated on Illumina’s X Ten we’re having to ask for “10X on X Ten” or “an X-Ten 10X genome” – I get them mixed up in conversations and I know PIs and post-dos do too!) Multiple deleterious cancer mutations, including the known driver genes TP53 and NRAS, five rearrangements and 26 copy-number variants were found . The most interesting result presented was a C>T mutation in TP53 that causes a deleterious nonsynonymous R213Q substitution, confirmed in the LinkedRead data as being on one haplotype. The other haplotype was shown to be deleted in the same region leading to LOH, with the only copy present having the TP53 C>T mutation resulting in a single but inactvated copy of TP53. This phased cancer genome was produced from 1ng of ~50kb DNA, from a sample with 70% tumour purity – this is pretty close to many samples that people are collecting, but the careful reporting of this kind of information is going to be vital as we understand which samples might sensibly be run on the 10X Genomics tech, and which we should leave for now.
A previously validated EML4-ALK translocation was detected in lung cancer cell line NCI-H2228. To target the exome for phasing 10X Genomics and Agilent have partnered on a modified capture panel that includes baits designed to target the introns and improve pull-down of the large genomic fragments. The sequencing of 200X was after removal of duplicates so this could be very deep sequencing indeed. However the 10X Genomics data revealed that this is not a simple inversion, but is a more complex with a deletion including exons 2–19 of ALK.
They discuss the Moleculo tech (actually refs 6-9 from the paper) from Illumina pointing out the main reasons that these methods are sub-optimal are the relatively large amount of DNA used and the relatively low number of partitions generated – both limiting how well the technology can be applied.
The authors conclude their discussion with the following statement “phased cancer genomes will provide new insight into the genomic alterations underlying tumor development and maintenance“. I think the next few months will see other papers being published confirming how useful the technology really is. And who knows how soon we might see a phasing panel specifically for DNA repair genes being used in the clinic for instance?
In this news and views article Jacob Kitzman (University of Michigan) describes the data from the Zheng et al paper (see above) in the same issue, and explores the impact it might have in the field. This paper clearly describes the issue that clinicians want to understand: are both copies of a gene affected e.g. as in cystic fibrosis, where two mutations, one on each allele knock out both copies of the CTFR gene, or if the same haplotype is hit twice with mutations in cis.
He suggests how other methods might be improved by the use of 10X phasing technology including metagenomics (we’re trying this with a collaborator), and for phasing cDNA to analyse transcriptomes more deeply with regards splice isoform diversity.
One of the questions Kitzman poses is “Whether the 10X Genomics platform will be widely adopted may depend as much on its cost above and beyond standard whole-genome shotgun sequencing as on its technical merit.” The papers above are showing just how useful the 10X Genomics tech is turing out to be…but as I said at the start of this post this list is going to grow rapidly; and this list will only be updated sporadically!







{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave A Comment