In this post I am going to outline the advice I give to my users.
The low diversity problem: The technology does suffer from a low diversity problem that has been explained over at Pathogenomics. They describe this “fly in the ointment” for Illumina users sequencing amplicons and point to a SEQanswers thread suggesting some work arounds e.g. spike in lots of PhiX. The main problem derives from the need to find distinct clusters on the images, this is done by HiSeq Control Software (HCS) on the fly. The first step is template generation which defines the X,Y coordinates of each cluster on a tile and is the reference for everything else. Template generation currently uses the first four cycles (but can be configured to use more) and this data is analysed after the fourth cycle is complete which users will see as a pause by the instrument. Clusters are found in each of the 16 images (A, C, G & T for four cycles). A Golden Cycle (g) is determined as the one with the most clusters in A&C, this is used as the reference for everything else. By comparing merged A&Cg clusters with A&C and G&T in all cycles a two-colour map of clusters is formed that distinguishes broad clusters from closely neighbouring but distinct ones. However all though there are four bases Illumina instruments only have two lasers, red (A&C) and green (G&T, which I remember by thinking of the colour of a Gordon’s gin bottle, green for G&T!) and in each cycle at least one of two nucleotides for each colour channel needs to be read to ensure proper image registration.
See technote_rta_theory_operations.pdf for more detail.
The causes of index read failures: If the index read is unbalanced due to poor mixing or low numbers of indexes then registration can fail due to the low diversity. The balance and number of libraries in a multiplex-pool impacts diversity and low diversity means too many clusters fail dempultiplexing and are thrown away. Illumina provide advice in their sample prep guides (e.g. TruSeq DNA PCR-Free Sample Preparation Guide (15036187 A)) on how best to quantify libraries before pooling and also on how to safely create low-plexity pools (more than 12 is usually safe). I’ll now address both of these; Quantifying & Normalising and Deciding Index Plexity in a little more detail.
Quantifying & Normalising: Simply use qPCR quantification to get the very best estimate of nM concentration. Bioanalyser, QuBit and Nanodrop all work but can be very inaccurate when compared to qPCR, it all depends on your libraries. These other methods also quantify molecules that cannot form clusters, such as molecules without both adapters, oligos and nucleotide and can significantly over- or under-estimate pM concentration. A very good QC document is one I found from an Illumina Aisa/Pacific meeting.
Illumina recommends the use of Kapa’s KK4824 Library Quantification Kit – Illumina/Universal kit, although now they are selling the Eco systems and their own NuPCR I’d expect an Illumina kit to come along any time soon.
KAPA’s is a SYBR-based real-time PCR assay that uses two primers complementary to the ends of TruSeq, Nextera and other library types. During PCR only amplifiable molecules contribute to the CT reading and so the assay is more robust than spectropohotometry or fluorimetry. The method is not without its drawbacks but if care is taken with preparation of the samples and controls then very accurate results are possible.
- Pipette large volumes – we use a 1:99ul dilution followed by serial 10:90ul dilutions to create a 1:100, 1:1000, 1:10,000 and 1:100,000.
- Replicate the dilutions – we make three independent serial dilutions.
- Use the controls – we aliquot controls to avoid freeze/thaw.
- Include NTC’s – vital if you want to see contamination, add them at different stages to see where contamination is coming from.
Two words of warning. Firstly total fragment length is required not just the insert size, don’t forget the adapters add another 120-130bp. Secondly, if you are using Illumina’s TruSeq PCR-free kits then do not use the Bioanalyser to estimate library size as it significantly over-estimates fragment length due to “the presence of certain structural features which would normally be removed if a subsequent PCR-enrichment step were performed”. The Illumina sample prep guide has a nice figure illustrating this comparing Bioanalyser and insert size distribution determined by alignment, and suggests using 470bp for 350bp libraries or 670bp for 550bp libraries in the calculations after Kapa qPCR.
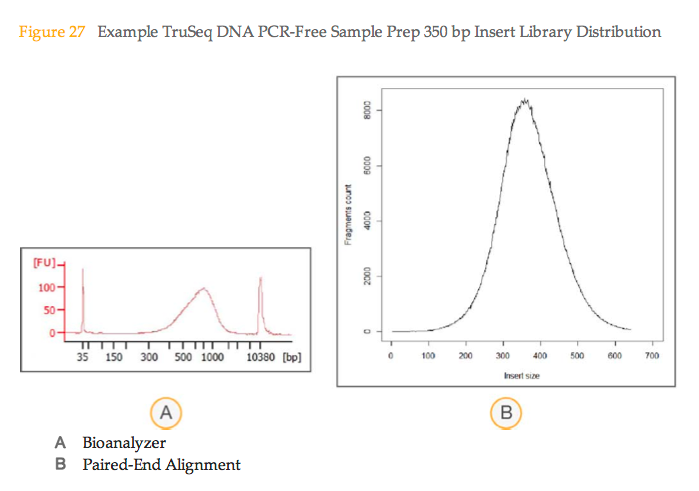
Figure 27 from Illumina’s TruSeq PCR-free guide
Improving qPCR: A big problem with qPCR as currently implemented is the need to know library size to calculate pM concentration. The assay uses an intercalating dye and is effectively measuring the amount of DNA present rather than the number of molecules. Fragment length is used to calculate number of molecules and if this is wrong then quantitation will also be inaccurate. Ideally we would not bother with the Bioanalyser but we need to understand insert size, this potentially doubles the work we need to do for QT of a library.
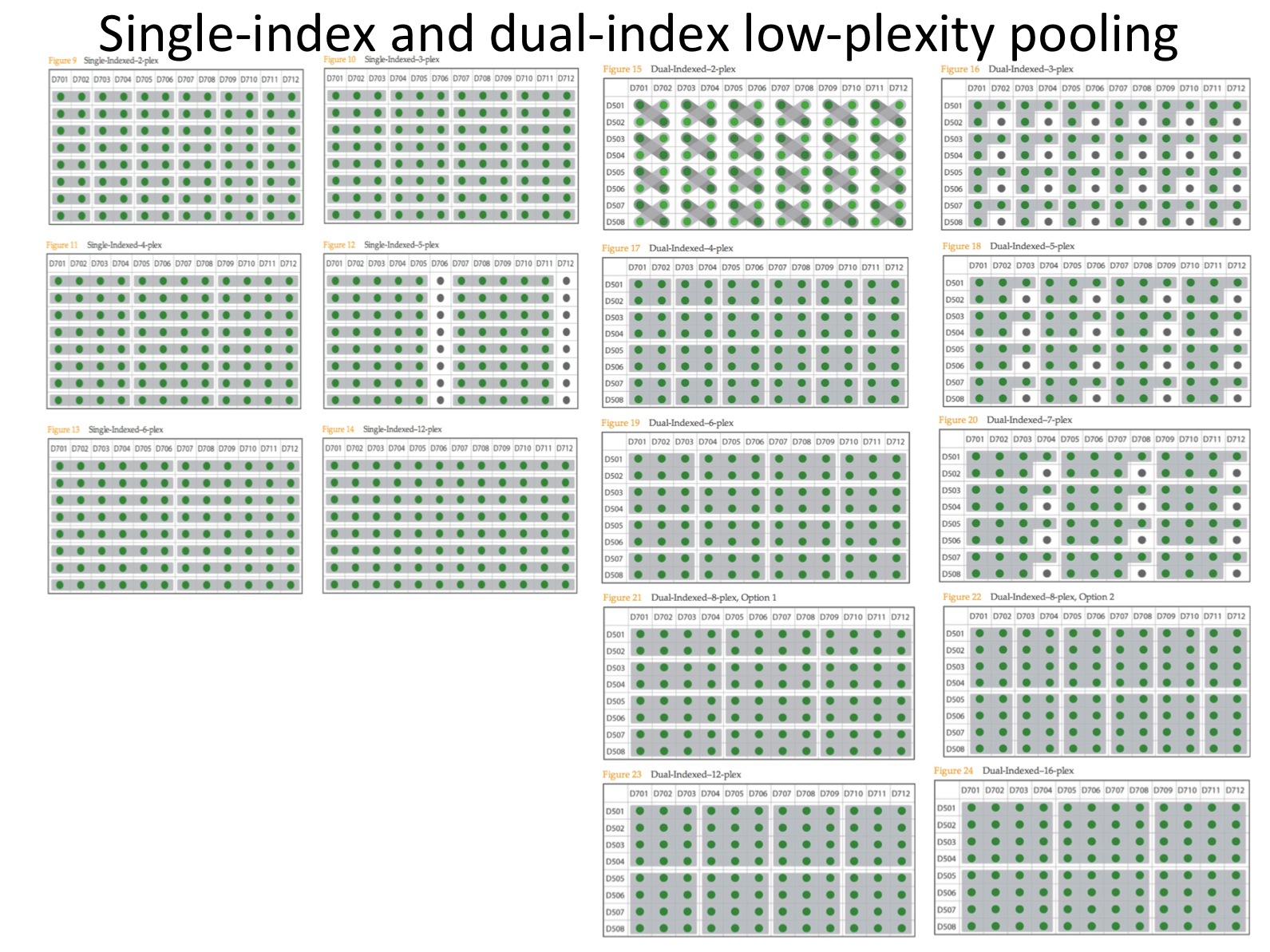 |
| Illumina’s low-plexity pooling guidelines. Simples! |
An added benefit of large “super-pools” being sequenced across multiple lanes and/or flowcells is that failures in the sequencing of any kind can be tolerated as long as most of the data is generated. IF a tumour:normal pair are sequenced as single lanes and one lane fails then no analysis can be performed. If the same pair are indexed and mixed with 7 other pairs and one lane on a flowcell fails copy number analysis would be hardly affected at all. The same principle applies to pretty much any method.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The lack of progress from Illumina on this 'low diversity' issue has surprised me given the importance of amplicon sequencing. I wonder if it's because they are focusing all of their efforts on the upcoming ordered arrays, as I assume this would solve the issue since each cluster location will be known.
Work is being done on this. Stay tuned…
Ooh… is Jay Flatley reading Core Genomics?
Interesting article. I've looked around your site and gives me some ideas of reading outside my usual comfort zone.
I've put together a website linked below that has been put together to share bioinformatics tutorials and create a dynamic learning environment that does not become dated, PDF contributions welcome and there are four core tutorials available. We would be interested to get some feedback form you and anything that you could post to educate.
http://elvis.ccc.cranfield.ac.uk/CUBELP/faces/login-page.xhtml