CirSeq
Identification of Low-Abundance RNA Viruses with Circular Sequencin
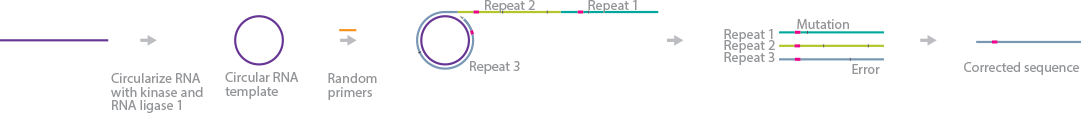
CirSeq accurately identifies ultra-rare and low-frequency genetic variants in RNA viruses. The method uses a unique step for circularization of fragmented viral RNAs, followed by rolling-circle RT (Acevedo et al., 2014) (Acevedo et al., 2014). CirSeq corrects for mutations introduced during its amplification steps by aligning the tandem-repeat sequences with each other and excluding those reads using informatics tools.
First, single-stranded RNAs are fragmented using Zn2+ and size-selected to no more than one-third of the sequencing read length. Next, they are circularized and reverse-transcribed using random primers. Rolling-circle RT is used to generate tandem-repeat cDNA strands. The first-strand cDNAs are amplified, generating double-stranded cDNAs, followed by end repair, poly(A) tailing, and adapter ligation. The cDNA libraries are ready for sequencing.
Advantages:
- Detects ultra-rare and low-frequency genetic variants in RNA viruses
- Rolling-circle RT creates tandem-repeat cDNAs that can be used to correct artificial mutations
- Error rates reported are far below those observed using standard RNA virus sequencing methods
Disadvantages:
- Whole process takes ~5 days
- Unsuitable for sequencing clinical isolates, because it needs large quantities of purified viral RNAs
- Not applicable for de novo sequencing of viral RNAs
Reagents:
Illumina Library prep and Array Kit Selector
Reviews:
Posada-Cespedes S., Seifert D. and Beerenwinkel N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res. 2016;
Andino R. and Domingo E. Viral quasispecies. Virology. 2015;479-480C:46-51
Gordon A. J., Satory D., Halliday J. A. and Herman C. Lost in transcription: transient errors in information transfer. Curr Opin Microbiol. 2015;24C:80-87
References:
Acevedo A., Brodsky L. and Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686-690
Wang K., Ma Q., Jiang L., et al. Ultra-precise detection of mutations by droplet-based amplification of circularized DNA. BMC Genomics. 2016;17:214
Related
History: CirSeq
Revision by sbrumpton on 2017-06-21 07:50:22 - Show/Hide
Identification of Low-Abundance RNA Viruses with Circular Sequencin
CirSeq accurately identifies ultra-rare and low-frequency genetic variants in RNA viruses. The method uses a unique step for circularization of fragmented viral RNAs, followed by rolling-circle RT (Acevedo et al., 2014) (Acevedo et al., 2014). CirSeq corrects for mutations introduced during its amplification steps by aligning the tandem-repeat sequences with each other and excluding those reads using informatics tools.
First, single-stranded RNAs are fragmented using Zn2+ and size-selected to no more than one-third of the sequencing read length. Next, they are circularized and reverse-transcribed using random primers. Rolling-circle RT is used to generate tandem-repeat cDNA strands. The first-strand cDNAs are amplified, generating double-stranded cDNAs, followed by end repair, poly(A) tailing, and adapter ligation. The cDNA libraries are ready for sequencing.
Advantages:- Detects ultra-rare and low-frequency genetic variants in RNA viruses
- Rolling-circle RT creates tandem-repeat cDNAs that can be used to correct artificial mutations
- Error rates reported are far below those observed using standard RNA virus sequencing methods
Disadvantages:- Whole process takes ~5 days
- Unsuitable for sequencing clinical isolates, because it needs large quantities of purified viral RNAs
- Not applicable for de novo sequencing of viral RNAs
Reagents:Illumina Library prep and Array Kit SelectorReviews:Posada-Cespedes S., Seifert D. and Beerenwinkel N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res. 2016;Andino R. and Domingo E. Viral quasispecies. Virology. 2015;479-480C:46-51Gordon A. J., Satory D., Halliday J. A. and Herman C. Lost in transcription: transient errors in information transfer. Curr Opin Microbiol. 2015;24C:80-87References:Acevedo A., Brodsky L. and Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686-690Wang K., Ma Q., Jiang L., et al. Ultra-precise detection of mutations by droplet-based amplification of circularized DNA. BMC Genomics. 2016;17:214